Synthesis of the natural herbicide δ-aminolevulinic acid from cellulose-derived 5-(chloromethyl)furfural
Mark
Mascal
* and
Saikat
Dutta
Department of Chemistry, University of California Davis, 1 Shields Avenue, Davis, CA 95616, USA. E-mail: mascal@chem.ucdavis.edu; Fax: 530-752-8995; Tel: 530-754-5373
First published on 6th December 2010
Abstract
Cellulose-derived 5-(chloromethyl)furfural is converted into δ-aminolevulinic acid in three chemical steps involving conversion to 5-(azidomethyl)furfural, photooxidation, and catalytic hydrogenation in 68% overall yield. δ-Aminolevulinic acid is a natural product with important agrochemical and pharmaceutical applications.
Having recently demonstrated that 5-(chloromethylfurfural) (CMF) 1 can be derived in a single step from either sugars, cellulose, or raw cellulosic biomass in yields between 80–90% of the theoretical,1 it is useful to describe the applications and markets addressed by this renewable platform chemical. Generally speaking, CMF 1 has two primary derivative manifolds; a) furanic, in which the furan ring remains intact, for example in the hydrolysis or alcoholysis of 1 to give hydroxy- or alkoxymethylfurfurals 2, and b) levulinic, wherein the carbonyl group is cleaved and the ring is opened to give levulinic and formic acid-based products 3 and 4 (Scheme 1).2 Herein, we describe an oxidative reaction pathway for 1 which enables a concise, green synthesis of the natural herbicide, insecticide, and photodynamic therapy drug δ-aminolevulinic acid (ALA).
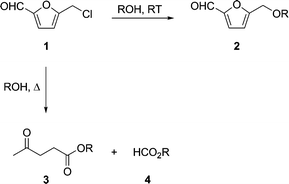 | ||
| Scheme 1 Derivatization routes for CMF 1. | ||
Considering the derivatization of 1, the natural focus is on the substituents. Thus, the aldehyde group could undergo a change in oxidation state, to the carboxylic acid, alcohol, or even methyl group, or it could participate in condensation reactions with nucleophiles, leading for example to carbon–carbon or carbon–nitrogen bond formation. The chloromethyl group on the other hand could be substituted with a wide range of carbon and heteroatom nucleophiles. Perhaps a less apparent point of derivatization is the furan ring itself, even though these are well known to undergo [4 + 2] cycloaddition reactions with a range of dienophiles.3 One potentially attractive transformation is the addition of singlet oxygen across the ring to give a 1,2,4-trioxolane, which is known in the case of furfural to decompose into the corresponding butenolide.4 The application of this chemistry to 5-substituted furfurals leads ultimately to α,β-unsaturated γ-oxo acids 7, a potentially useful class of furan derivatives (Scheme 2). In fact, the approach has been used by two groups in the synthesis of ALA. Thus, Descotes and co-workers converted 5-(hydroxymethyl) furfural (HMF) 2 (R = H) into the 5-acetamido derivative using Ritter chemistry. The furan ring was oxidized with singlet oxygen to give 6 (R = CH2NHAc) which was reduced with Zn–AcOH and the amide hydrolyzed to conclude a 4-step route to ALA in 18% overall yield.5 Takeya and co-workers described a similar 4-step synthesis of ALA starting from furfurylamine, which was protected first as the phthalimide and converted with singlet oxygen directly into 5-phthalimido-β-acetylacrylic acid 7 (R = CH2N(CO)2C6H4). This material was hydrogenated and the imide group hydrolyzed to give ALA in a 50% overall yield.6 The furfurylamine starting material can be sourced by the reductive amination of furfural.7–9
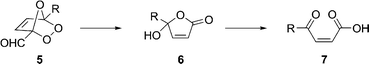 | ||
| Scheme 2 Transformations of furan-derived 1,2,4-trioxolanes. | ||
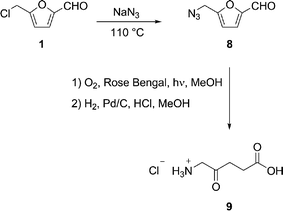 | ||
| Scheme 3 Synthesis of ALA 9 from 1. | ||
The problem with both of the above approaches is their reliance on protecting groups, which presents a considerable disadvantage in terms of atom economy. Clearly, an unprotected amine would interfere with the oxidation reaction, but we reasoned that if an azide group were used as the amine precursor, both it and the double bond in intermediate 7 could be reduced at the same time, saturating the chain and unmasking the NH2 group in a single operation. We therefore recognized the potential of using biomass-derived CMF 1, with its reactive chloro group, as the starting material for a singlet oxygen-mediated synthesis of ALA. Thus, 5-(azidomethyl) furfural 8 was prepared in 92% yield from a neat mixture of 1 and NaN3 and then irradiated with oxygen in the presence of Rose Bengal to give a mixture of 6 and 7 (R = CH2N3). These products were not isolated but the mixture was directly hydrogenated to give ALA 9 (as the stable hydrochloride salt) in 3 steps and an overall yield of 68% from 1 (Scheme 3).10 If this result is superimposed on previously reported yields of 1 from biomass sources1 the result would be 61, 57, and 55% isolated chemical yield of ALA 9 from sucrose, cellulose, and corn stover, respectively.
ALA 9 is an intermediate in the biosynthesis of tetrapyrrole, which is the core structure of porphyrins and heme. It is a compound of current interest as both a herbicide and insecticide due to the fact that it is highly active, biodegradable, and nontoxic to humans and animals.11 It has also been proposed as a plant growth regulator,12 and increases salt and cold temperature tolerance in crops.13 Presently, ALA is employed as a sensitizer in photodynamic therapy for cancer and dermatological disorders.14 The reason for the limited adoption of ALA in agricultural practice has been its poor availability and therefore high price. It can be produced microbially,13 but this is slow and expensive, and generally not well suited to production on an agrochemical scale. Other chemical routes vialevulinic acid itself have generally suffered from poor yields.15–19 We suggest that the efficient, renewable approach to the synthesis of ALA 9 described here will enable broader applications of this valuable natural product.
Experimental section
5-(Azidomethyl)furfural 8
A mixture of 5-(chloromethylfurfural) 1 (2.07 g, 14.3 mmol) and sodium azide (1.04 g, 16.0 mmol) was heated at 110 °C for 3 h under argon with stirring. The reaction was cooled to room temperature and CH2Cl2 (100 mL) was added. The mixture was filtered through a plug of silica gel and the solvent was evaporated to give 8 (1.99 g, 92%) as a pale yellow oil. 1H NMR (CDCl3, 300 MHz) 9.64 (1H, s), 7.23 (1H, d, J = 3.4 Hz), 6.56 (1H, d, J = 3.4 Hz), 4.42 (2H, s); 13C NMR (CDCl3, 75 MHz) 177.8, 155.5, 152.9, 122.3, 111.7, 47.0; IR (neat): νmax 2920, 2850, 2096, 1673, 1519, 1468, 1401, 1330, 1271, 1232, 1191, 1022 cm−1; HRMS (ESI): calculated for C6H6N3O2: [M+H]+ 152.0454; found: 152.0451.5-Aminolevulinic acid hydrochloride 9
A mixture of 8 (0.250 g, 1.65 mmol) and Rose Bengal (10 mg) in dry methanol (20 mL) was irradiated with a halogen lamp (Sylvania 250 W) for 6 h, during which a gentle stream of oxygen was passed through the solution. The reaction mixture was kept at 20 °C by use of an ice-water bath. Concentrated HCl (1.0 mL) and 10% Pd/C (60 mg) were then added and the mixture was shaken under 2 atm H2 for 6 h. The reaction was filtered to remove the catalyst and the resulting colorless solution was evaporated to give a beige solid. The crude product was dissolved in methanol (10 mL) and re-precipitated by the addition of ether to give the pure product (0.206 g, 74%), mp 143–146 °C (lit. 144–147 °C).201H NMR (D2O, 300 MHz) 4.01 (2H, s), 2.77 (2H, t, J = 6.1 Hz), 2.59 (2H, t, J = 6.1 Hz); 13C NMR (D2O, 75 MHz) 204.0, 176.7, 47.2, 34.5, 27.8.Acknowledgements
This research was supported by the US National Science Foundation, grant CBET 0932391.References
- M. Mascal and E. B. Nikitin, ChemSusChem, 2009, 2, 859 CrossRef CAS.
- M. Mascal and E. B. Nikitin, Green Chem., 2010, 12, 370 RSC.
- C. O. Kappe, S. S. Murphree and A. Padwa, Tetrahedron, 1997, 53, 14179 CrossRef CAS.
- G. O. Schenck, Justus Liebigs Ann. Chem., 1953, 584, 156 CrossRef CAS.
- L. Cottier, G. Descotes, L. Eymard and K. Rapp, Synthesis, 1995, 303 CrossRef CAS.
- H. Takeya, H. Ueki, S. Miyanari, T. Shimizu and M. Kojima, J. Photochem. Photobiol., A, 1996, 94, 167 CrossRef CAS.
- M. Wu and Y. Xu, Huaxue Shiji, 1981, 6, 379.
- T. Ayusawa, S. Mori, T. Aoki and R. Hamana, Chem. Abstr., 104, 50784 Search PubMed , Japanese Patent 60146885.
- S. Mori, R. Hamana, T. Aoki and T. Ayusawa, Chem. Abstr., 106, 18343 Search PubMed , Japanese Patent 61134384.
- The 1H and 13C NMR chemical shifts of 1 are consistent with reported values in the literature: P. B. Shrestha-Dawadi and J. Lugtenburg, Eur. J. Org. Chem., 2003, 4654 Search PubMed.
- Review: C. Sasikala, C. V. Ramana and P. R. Rao, Biotechnol. Prog., 1994, 10, 451 Search PubMed.
- K. Watanabe, T. Tanaka, Y. Hotta, H. Kuramochi and Y. Takeuchi, Plant Growth Regul., 2000, 32, 99 CAS.
- Review: K. Sasaki, M. Watanabe, T. Tanaka and T. Tanaka, Appl. Microbiol. Biotechnol., 2002, 58, 23 Search PubMed.
- Review: H. Fukuda, A. Casas and A. Batlle, Int. J. Biochem. Cell Biol., 2005, 37, 272 Search PubMed.
- Review: L. Moens, ACS Symp. Ser., 2001, 784, 37 Search PubMed (Chemicals and Materials from Renewable Resources).
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227 CrossRef.
- S. F. MacDonald, Can. J. Chem., 1974, 52, 3257 CrossRef CAS.
- H.-J. Ha, S.-K. Lee, Y.-J. Ha and J.-W. Park, Synth. Commun., 1994, 24, 2557 CAS.
- R. Vallinayagam, H. Bertschy, Y. Berger, V. Wenger and R. Neier, Synthesis, 2007, 3731 CAS.
- A. A. Marei and R. A. Raphael, J. Chem. Soc., 1958, 2624 RSC.
| This journal is © The Royal Society of Chemistry 2011 |