Efficient preparation of β-D-glucosyl and β-D-mannosyl ureas and other N-glucosides in carbohydrate melts†
Carolin
Ruß
a,
Florian
Ilgen
a,
Christian
Reil
a,
Claudia
Luff
a,
Alireza
Haji Begli
b and
Burkhard
König
*a
aInstitute of Organic Chemistry, University of Regensburg, Universitätsstraße 31, 93040, Regensburg, Germany. E-mail: burkhard.koenig@chemie.uni-regensburg.de; Fax: +49 9419 4317 17; Tel: +49 9419 4345 66
bSÜDZUCKER AG Mannheim/Ochsenfurt Zentralabteilung Forschung, Entwicklung, Services (ZAFES), Wormser Straße 11, D-67283, Obrigheim, Germany
First published on 6th December 2010
Abstract
Sugar melts or solvent-free systems have been used to react simple unprotected hexoses at the C-1 atom with urea and urea derivatives to sugar-ureides by acid catalysis and with short reaction times. In one step, β-D-glucosyl- and β-D-mannosyl urea 2a/b were obtained in high yields. D-Galactose 6, N-acetyl-D-glucosamine 7, L-rhamnose 8 and 2-deoxy-D-glucose 9 were converted likewise to glycosyl ureas. Additionally, urea-related nucleophiles were investigated as melt components. N,N′-Ethylene urea 15, N,N′-allylurea 16 and ethyl carbamate 18 were β-selectively converted with D-glucose in good yield, giving the corresponding N-glycosides. Under these conditions, however, the condensation product with N-octylurea 17 was not accessible.
Introduction
In the 21st century, the utilisation of renewable raw material will gain significant importance in the industrial conversion of chemicals. This fact is a consequence of diminishing fossil fuel reserves, which will urge the development of new methodologies to make use of sustainable sources for chemical production in the near future.1 Since biomass is renewable, abundant and distributed widely in nature, it is a promising alternative for the sustainable supply of valuable intermediates and platform chemicals to the chemical industry.2Carbohydrates form the main part of biomass with more than 75 wt%.3 They can be used directly for chemical conversion or after the hydrolysis of poly- and oligosaccharides to monosaccharides like D-glucose and D-fructose. Substitution at the most oxidised site in monosaccharides, the anomeric centre, gives access to important and prominent groups of glycosides. O-,4S-,5C-6 and N-7glycosides are examples in this group of C-1 substituted monosaccharides. A representative of N-glycosides is the stable class of glycosyl ureas. Glycosyl ureas are widely used in mixtures with phenol and water as adhesives with excellent properties. This formulation is important for the forest product industry, which is interested in reducing the phenol content of adhesives in construction materials and furniture due to the toxicity of phenol.8 Glycosylthymines can be prepared from glycosyl ureas, as described by Sano et al.9 Another important application of glycosyl ureas is their use as lyophilisation stabilisers for enzymes.10 Recently, Shoji et al. introduced a glycosyl urea based lectin adsorbent with a high and controllable adsorption capacity that can be manufactured conveniently.11 Structurally similar N-acyl-N′-β-glucopyranosyl ureas were identified as nanomolar inhibitors of rabbit muscle glycogen phosphorylase and might be applied in the therapy of type 2 diabetes mellitus.12N-Aryl-N′-β-glucopyranosyl ureas exhibit weaker binding to the glycogen phosphorylase than acyl derivatives.12a,13
The condensation products between aldoses and urea are obtained from acid catalysed reactions in water or water mixtures, and were first described for D-glucose by Schoorl et al. as early as 1900.14 After minor modifications to the original procedure, the synthesis of glycosyl ureas was improved by Benn and Jones, yielding 32% after 42 h with sulfuric acid as the catalyst.15 The best results so far have been obtained by Sano et al. using the ion exchanger Amberlite IR-120 (H-form) to obtain β-D-glycosyl urea in 53% chemical yield after 4 d at 75–80 °C.9 Higher yields could not be achieved without significantly longer reaction times (7–14 d).8
Modern and more versatile methods use the reaction of glycosyl isocyanates with amines to prepare glycosyl ureas. These and other important synthetic approaches towards carbohydrate-based ureas have been reviewed by Spanu and Ulgheri.16 A simple synthesis of α-glycosyl ureas has been developed by Bianchi et al.17
The reported methods for the preparation of β-D-glycosyl and β-D-mannosyl ureas suffer from moderate yields and long reaction times. An ideal method for the conversion of biomass into platform chemicals, however, is the use of highly concentrated systems featuring a high substrate concentration and high chemical yields. Such systems should allow efficient conversions and short reaction times.
Here, we report the application of carbohydrate urea melts developed in our working group18 with diverse Brønsted and Lewis acids as catalysts in aldose concentrations as high as 3 mol L−1. Using such a carbohydrate melt system, the reaction times are reduced, while the yields are significantly increased compared to previously reported systems (up to 78%). Apart from β-D-glucosyl urea 2a, β-D-mannosyl urea 2b was prepared in the highest yield reported (up to 81%) so far in the literature. Glycosyl urea formation was also observed for D-galactose 6, N-acetyl-D-glucosamin 7, L-rhamnose 8 and 2-deoxy-D-glucose 9. Moreover, we show that N,N′-ethylene urea 14, N,N′-allylurea 15 and ethyl carbamate add in the melt to the C-1 position of D-glucose.
Results and discussion
Formation of β-D-glucosyl urea in a carbohydrate melt
The acid catalysed condensation of D-glucose with urea in aqueous media by applying long reaction times was described by both Benn et al.15 and Sano et al.9 The stereochemistry at the anomeric centre was determined by Helm8 to be the β-form based on the 1H-NMR coupling constants of the two axial protons in the C-1 and C-2 positions. Typically, the anomeric effect favours the α-configuration in sugars with electronegative substituents in the C-1 position.19 Here, nitrogen has a lower electronegativity compared to oxygen and halogens, and thus contributes less to the anomeric stabilisation. Polar solvents are known to reduce the stabilisation at the anomeric centre. Both of these effects and steric hindrance account for the preferred β-glycoside configuration.The first results for the condensation reaction were obtained by using montmorillonite as the catalyst in a D-glucose/urea/NH4Cl melt (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7:1 wt/wt/wt). Montmorillonite, a phyllosilicate with Brønsted and Lewis acid character, was chosen as a catalyst because it is mild, non-toxic and could be recycled after the reaction, since it is a heterogeneous catalyst. After a 48 h reaction time at 80 °C, the reaction was analysed by 13C-NMR and showed a high conversion and selectivity. The resonance signal at the anomeric centre (92.3 ppm, d6-DMSO) disappeared completely and the only carbonyl resonance signal, detected at 158.0 ppm (d6-DMSO), indicated the selective formation of one isomer. The sample was analysed by mass spectrometry to confirm that the urea was selectively mono-glycosylated. The coupling constant in the 1H-NMR spectrum confirmed the β-configuration of the glycoside (Scheme 1).
:7:1 wt/wt/wt). Montmorillonite, a phyllosilicate with Brønsted and Lewis acid character, was chosen as a catalyst because it is mild, non-toxic and could be recycled after the reaction, since it is a heterogeneous catalyst. After a 48 h reaction time at 80 °C, the reaction was analysed by 13C-NMR and showed a high conversion and selectivity. The resonance signal at the anomeric centre (92.3 ppm, d6-DMSO) disappeared completely and the only carbonyl resonance signal, detected at 158.0 ppm (d6-DMSO), indicated the selective formation of one isomer. The sample was analysed by mass spectrometry to confirm that the urea was selectively mono-glycosylated. The coupling constant in the 1H-NMR spectrum confirmed the β-configuration of the glycoside (Scheme 1).
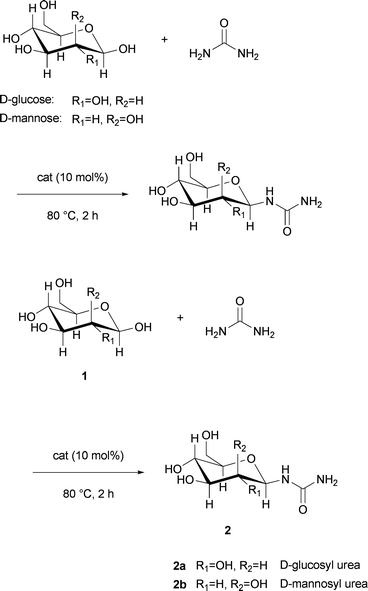 | ||
| Scheme 1 Acid catalysed formation of β-form condensation product 2a/b in the melt. | ||
After the initial experiment, several other catalysts were tested. The product yield was determined by HPLC using sucrose as an internal standard (Table 1).
The highest yield for glycoside 2a was 81% with Amberlyst 15 after 2 h at 80 °C, as determined by HPLC. para-Toluene sulfonic acid (p-TsOH) and FeCl3 yielded 37 and 27%, respectively. Montmorillonite and ZnCl2 displayed no catalytic reactivity. Additionally, the HPLC measurements showed that after 30 min, at least 60% of D-glucose 1a had been converted. After 2 h, about 8% sugar 1a could be detected, even in the absence of a catalyst. The discrepancy between the yield and the D-glucose consumption is mostly due to the formation of a second product, detected as a single peak next to the product peak in the HPLC chromatogram. The integrals of the HPLC signals were compared, which is possible for the nearly quantitative ELSD detector due to uniform responses. After a 15 min reaction time with Amberlyst 15, the intermediate was 10% less than the amount of glucosyl urea, whereas after 2 h, only 7% was present. A 6–7-fold amount of the second product compared to glucosyl urea 2a was found without catalyst after 15 min, and after 6 h, still in a 4-fold excess. Further LC-MS analysis proved that the unknown product exhibits a molecular weight of 240 g mol−1, which might correspond to intermediate 3 (Scheme 2), obtained by nucleophilic addition to C-1.
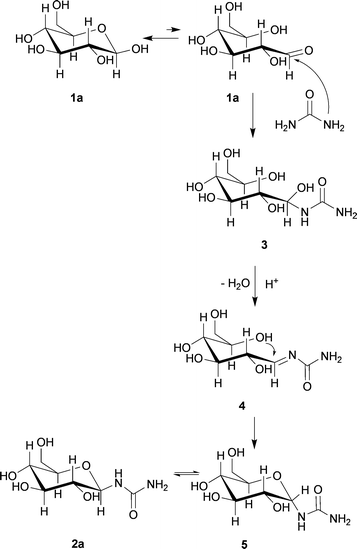 | ||
| Scheme 2 A suggested reaction mechanism for the reaction of D-glucose 1avia intermediate 3 in a sugar–urea–salt melt under acidic conditions. | ||
Formation of β-D-mannosyl urea in carbohydrate melt
Based on the successful conversion of D-glucose 1a, the epimer D-mannose 1b was tried to show the general applicability of acid catalysed condensation with urea in high concentration carbohydrate melts for different sugars (Scheme 1). Badawi reported an inefficient procedure in water with sulfuric acid as the catalyst and reaction times of up to 7 d. The yield of β-D-mannosyl urea 2b after 7 d was 12% after recrystallisation from MeOH.20 A β-configuration at the anomeric centre was established by optical rotation of the derivatives after a periodate reaction, which was compared with the value of derivatives of β-D-glycosyl urea 2a.In an initial study, a melt consisting of a D-mannose/urea/NH4Cl melt (3:7:1 wt/wt/wt) was stirred with Amberlyst 15 as the catalyst, and the purified product was analysed by NMR and mass spectrometry. 13C-NMR and NOE experiments (see the ESI†) confirmed the expected β-anomer as the reaction product, and the mass spectrometry analysis indicated that selective mono-condensation had taken place. Again, the reduced electronegativity and bulkiness of the urea moiety are the probable reason for the observed stereochemistry.
Quantification of sugar ureide 2b by HPLC showed that optimum yields with selected catalysts were achieved after a 1 h reaction time at 80 °C.
The best yields of condensation product 2b were obtained with FeCl3 (81%), Amberlyst 15 (75%) and p-TsOH (64%). Montmorillonite and ZnCl2 were also applied but showed less catalytic activity, and the determined yields remained below the other catalysts. In contrast to the condensation with monosaccharide 1a, only one product was found by HPLC.
With 1 and 2 h, respectively, for both β-D-mannosyl and β-D-glycosyl urea 2b/a, the reaction time could be significantly reduced compared to the previous literature.
A fructose/urea melt was reacted under acidic conditions (Amberlyst 15) at 80 °C for 24 h and formed a mixture of condensation products that could not be separated. We assume that the fructose/urea condensation products are present as furanose and pyranose, as well as in their α- and β-forms.
Reactions of further monosaccharides in carbohydrate–urea melts
To investigate the effect of stereochemistry on the reaction pathway and to enlarge the scope of application, four additional sugar/urea/NH4Cl melts (3:7:1 wt/wt/wt) were examined for their reaction under acidic conditions. In the depicted conformations, D-galactose 6 has an axial OH-group at the 4-position, N-acetyl-D-glucosamine 7 is substituted with a bulky and electron-withdrawing group at the 2-position, L-rhamnose 8 shows, like D-mannose, an axial OH-group at the 2-position and 2-deoxy-D-glucose 9 has the OH-group at the 2-position replaced by a hydrogen atom (Scheme 3).
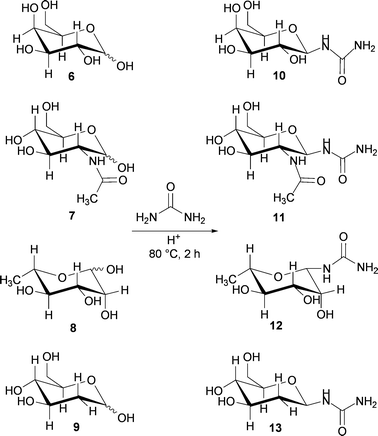 | ||
| Scheme 3 Glycosyl ureas 10–13 from the acid catalysed reaction of monosaccharides 6–9 in melts. | ||
After stirring the melts with acidic Amberlyst 15 at 80 °C for 2 h, reaction control by 13C-NMR showed that the 12 signals of the corresponding starting materials were reduced to 6 signals. As in the case of glucosyl- and mannosyl urea, the resonance signal for the anomeric centre disappeared and a new carbonyl resonance was detected at 160 ppm (D2O). Apparently, one anomer was selectively formed, presumably the β-anomer. HPLC-MS measurements and mass analysis confirmed for all the monosaccharides that only a single glycosyl urea was formed (see the ESI†). Additionally, we observed that more than 90% of the starting material was converted. Neither in the samples with catalyst nor in the samples without catalyst was an intermediate (cf.D-glucose 3) found.
Reactions of D-glucose with urea derivatives under solvent-free and acidic conditions
After efficient condensation of monosaccharides 1a/b and 6–9 with urea, different urea derivatives and nucleophiles with similar structures to urea were tested as melt components to form N-glucosides (Scheme 4).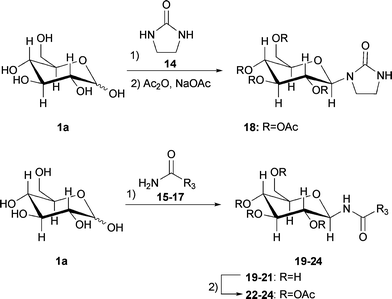 | ||
| Scheme 4 Acid catalysed formation of condensation products 19–24 in melts. | ||
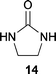
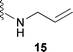
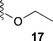
Therefore, the lowest melting point (eutectic point) was determined for mixtures of sugar 1a and one of the additive compounds 14–17 (Table 2). However, only cyclic N,N′-ethylene urea 14 showed a melting point depression and a clear melt was formed. In all other cases, the sugar could only be suspended in an excess of the melted component.
| Compound | R3 | Reaction time/h | T/°C | Yield (%) |
|---|---|---|---|---|
| 18 |
|
6 | 75 | 27 |
| 19, 22 |
|
2 | 85 | 60 |
| 20, 23 |

|
2–24 | 110 | — |
| 21, 24 |
|
4 | 70 | 73 |
The addition of Amberlyst 15 to a 1:1 (wt/wt) mixture of cyclic N,N′-ethylene urea 14 and D-glucose 1a with stirring of the melt for 6 h at 75 °C afforded 27% of pure N-D-glucosyl-N′-ethylene urea tetraacetate 18 after acetylation and purification by column chromatography or recrystallisation. A corresponding mixture of cyclic urea 14 with D-fructose showed the formation of the dehydration product HMF after stirring at 85 °C overnight with Amberlyst 15.
Although D-glucose 1a and N-allyl urea 15 do not form a clear melt, the addition of Amberlyst 15 to a suspension of D-glucose/N-allyl urea (2:1, mol/mol) at 85 °C catalysed the condensation of the urea derivative to the C-1 position of compound 1a. After a 2 h reaction time, sugar 1a was completely consumed and about 50% of the unprotected product N-D-glucosyl-N′-allyl urea 19 was formed (NMR estimation). Due to the amphiphilic character of the molecule, only analytical amounts of the unprotected form could be isolated. From the large 1H coupling constant (J = 9.1 Hz) between the H-atom of the anomeric centre and the H-atom of the adjacent carbon in the unprotected sugar, we infer that both atoms have an axial–axial configuration, thus the β-form is favoured. HPLC measurements confirmed the highest yield after 2 h. Acetylation and purification by column chromatography afforded 60% of pure product 22.
Sugar-based surfactants could be obtained by the condensation of long-chain alkyl ureas and saccharides. Procedures for the direct condensation of aldohexoses with such urea derivatives in solution are already published.21 Another strategy without protecting groups involves the use of D-glucosylamines and alkyl isocyanates.21b,22
Under solvent-free conditions in a suspension of melted N-octylurea 16 and D-glucose 1a with varying acidic catalysts, a number of products were formed. The low reactivity of the long-chain alkyl ureas might be explained by intramolecular hydrogen bonding between the alkyl ureas.23 Furthermore, many products were generated, presumably due to Maillard-like reactions or caramelization at such high reaction temperatures.
Finally, an N-glucoside of ethyl carbamate 17 with D-glucose 1a was synthesized. Carbamates are structurally related to ureas, also called carbamides. They are established as protecting groups for amine groups and can be cleaved by various chemical and enzymatic methods. Tetraacetyl-D-glucosylethylurethane was firstly reported by Helferich et al. and synthesized by the reaction of tetraacetyl-D-glucosamine with ethyl chloroformate in dry pyridine.24 Another method developed by Sarap et al. involves the synthesis of tetra-O-acetyl-β-D-glucosyl isocyanate and its reaction with ethanol.25 In an initial screening of acid catalysts, FeCl3 was identified as the catalyst with the highest conversion. In a suspension of ethyl carbamate 17 and D-glucose 1a (2:1, mol/mol) with 10 mol% of the catalyst after a 5 h reaction time at 70 °C, 73% of the product was found after acetylation and purification. Only analytical amounts of unprotected N-D-glucosyl-ethyl carbamate 21 could be isolated by our means. In this case, again, the β-anomer is the only obtained isomer, which was confirmed by the large coupling constant (J = 9.3 Hz) in the 1H-NMR spectrum.
Experimental
General
All chemicals for syntheses were used as received without further purification. N-Octylurea was prepared according to the procedure of Kehm.26IR spectra were recorded using a Bio-Rad FT-IR-FTS 155 spectrometer. Melting points were determined by an Optimelt MPA 100 apparatus from Stanford Research Systems.NMR spectroscopy
NMR spectra were recorded on a Bruker Avance 600 (T = 300 K) instrument. The spectra were referenced against the internal NMR-solvent standard, and chemical shifts are reported in ppm.HPLC measurements
:5:5 as the eluent. The column temperature was 40 °C, the injection volume was 0.1 μL, while a flow rate of 0.3 mL min−1 and sucrose as an internal standard were used. The system was run with ChemStation for LC 3D Systems Rev. B.03.02 software.
:5:5 as the eluent. The column temperature was 25 °C, the injection volume was 0.5 μL, while a flow rate of 0.3 mL min−1 and sucrose as an internal standard were used. The system was run with ChemStation for LC 3D Systems Rev. B.03.02 software.
Typical procedure for the preparation of β-D-glucosyl urea 2a
D-Glucose (0.6 g, 3.3 mmol), urea (1.4 g, 26.7 mmol) and NH4Cl (0.2 g, 3.7 mmol) were melted in a 25 mL reaction flask at 80 °C until a clear melt was formed. Amberlyst (0.2 g) was added and the reaction stirred for 2 h at that temperature. After the reaction was finished, water was added to the still warm melt and the catalyst was filtered off. After the removal of water, the brownish solid was twice recrystallised from MeOH to give pure β-D-glycosyl urea as white crystals (0.47 g, 64%) (for characterisation data see ref. 8).β-D-Mannosyl urea 2b
D-Mannose (0.6 g, 3.3 mmol), urea (1.4 g, 26.7 mmol) and NH4Cl (0.2 g, 3.7 mmol) were melted in a 25 mL reaction flask at 80 °C until a clear melt was formed. Amberlyst 15 (0.2 g) was added and the reaction stirred for 1 h at that temperature. After the reaction was finished, water was added to the still warm melt and the catalyst was filtered off. After the removal of water, the brownish solid was twice recrystallised from MeOH to give a mixture of β-D-mannosyl urea and urea. The urea was degraded by urease in an aqueous solution, the urease filtered off, and after freeze-drying pure β-D-mannosyl urea was obtained as a white powder (0.53 g, 72%).1 H-NMR (600 MHz, DMSO- d 6 : δ [ppm] = 2.97–3.03 (m, 1 H), 3.24–3.34 (m, 2 H), 3.36–3.43 (m, 1 H), 3.50–3.52 (m, 1 H), 3.59–3.65 (m, 1 H), 4.41 (t, J = 6.0 Hz, 1 OH), 4.67 d (d, J = 5.0 Hz, 1 OH), 4.76 (d, J = 5.4 Hz, 1 OH), 4.80 (m, 1 H), 4.83 (d, J = 5.4 Hz, 1 OH), 5.84 (s, NH2), 6.47 (s, NH); 13C-NMR (150 MHz, DMSO-d6): δ [ppm] = 61.41, 66.88, 71.22, 74.39, 78.30, 78.46, 157.53; FT-IR (ATR): ν [cm−1] = 3334, 3244, 2942, 2358, 1663, 1614, 1528, 1446, 1411, 1377, 1200, 1140, 1076, 1047, 1024, 958, 863, 801,614, 539; m.p.: 178 °C; LSI-MS (glycerol): m/z (%) = 223.1 (100) [MH+], 315.3 (43) [MH+ + glycerol]; LSI-MS: calc.: 223.0930, found: 223.0933.
Conclusions
In conclusion, we have shown that carbohydrate–urea melts are suitable reaction media to synthesize N-glycosides efficiently in high yields (up to 81%) under mild reaction conditions and high concentrations. The readily available starting materials consist mainly of renewables and cheap bulk chemicals. In a one-step reaction and without the need for protecting groups, the β-anomer was formed selectively. Our data imply that the reaction of D-glucose may proceed via an intermediate O,N-hemiacetal, formed by the addition of the nucleophile to C-1. Likewise, D-glucose, D-mannose, D-galactose, N-acetyl-D-glucosamin, L-rhamnose and 2-deoxy-D-glucose were converted, and the scope of the melt condensation reaction includes N-substituted ureas and carbamates. The reported glycosyl urea synthesis reported here is superior to previously reported procedures as it is very simple and efficient.Acknowledgements
We thank the Südzucker AG and the Fachagentur Nachwachsende Rohstoffe for financial support.References
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef CAS.
- Y. Roman-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef CAS.
- D. Peters, Chem. Ing. Tech., 2006, 78, 229–238 CrossRef CAS.
- R. R. Schmidt and W. Kinzy, in Advances in Carbohydrate Chemistry and Biochemistry, ed. H. Derek, Academic Press, 1994, vol. 50, pp. 21–123 Search PubMed.
- J. D. Wander and D. Horton, in Advances in Carbohydrate Chemistry and Biochemistry, ed. R. S. Tipson, Academic Press, 1976, vol. 32, pp. 15–123 Search PubMed.
- (a) M. D. Lewis, J. K. Cha and Y. Kishi, J. Am. Chem. Soc., 1982, 104, 4976–4978 CrossRef CAS; (b) I. Paterson and L. E. Keown, Tetrahedron Lett., 1997, 38, 5727–5730 CrossRef CAS.
- W. Pigman, D. Horton and A. Herp, The Carbohydrates: Chemistry and Biochemistry, Academic Press, New York, 2nd edn, 1970 Search PubMed.
- R. F. Helm, J. J. Karchesy and D. F. Barofsky, Carbohydr. Res., 1989, 189, 103–112 CrossRef CAS.
- T. Naito, M. Hirata, T. Kawakami and M. Sano, Chem. Pharm. Bull., 1961, 9, 703–708 CAS.
- H. Hayade and W. Tsugawa, JP Pat., 2002-64466, 2003261591, 2003 Search PubMed.
- S. Nagaoka, T. Sato, H. Ihara, S. Ishihara and G. Maruyama, JP Pat., 2003-413746, 2004203870, 2004 Search PubMed.
- (a) L. Somsak, K. Czifrak, M. Toth, E. Bokor, E. D. Chrysina, K. M. Alexacou, J. M. Hayes, C. Tiraidis, E. Lazoura, D. D. Leonidas, S. E. Zographos and N. G. Oikonomakos, Curr. Med. Chem., 2008, 15, 2933–2983 CrossRef CAS; (b) L. Somsák, N. Felföldi, B. Kónya, C. Hüse, K. Telepó, É. Bokor and K. Czifrák, Carbohydr. Res., 2008, 343, 2083–2093 CrossRef CAS.
- N. Felföldi, M. Tóth, E. D. Chrysina, M.-D. Charavgi, K.-M. Alexacou and L. Somsák, Carbohydr. Res., 2010, 345, 208–213 CrossRef CAS.
- (a) N. Schoorl, Rec. trav. chim. Pays-Bas, 1901, 19, 398–400; (b) N. Schoorl, Rec. trav. chim. Pays-Bas, 1903, 22, 31–77.
- M. H. Benn and A. S. Jones, J. Chem. Soc., 1960, 3837–3841 RSC.
- P. Spanu and F. Ulgheri, Curr. Org. Chem., 2008, 12, 1071–1092 CrossRef CAS.
- A. Bianchi, D. Ferrario and A. Bernardi, Carbohydr. Res., 2006, 341, 1438–1446 CrossRef CAS.
- (a) F. Ilgen and B. Konig, Green Chem., 2009, 11, 848–854 RSC; (b) F. Ilgen, D. Ott, D. Kralisch, C. Reil, A. Palmberger and B. Koenig, Green Chem., 2009, 11, 1948–1954 RSC; (c) G. Imperato, E. Eibler, J. Niedermaier and B. Konig, Chem. Commun., 2005, 1170–1172 RSC; (d) G. Imperato, S. Hoger, D. Lenoir and B. Konig, Green Chem., 2006, 8, 1051–1055 RSC; (e) G. Imperato, B. König and C. Chiappe, Eur. J. Org. Chem., 2007, 1049–1058 CrossRef CAS; (f) G. Imperato, R. Vasold and B. König, Adv. Synth. Catal., 2006, 348, 2243–2247 CrossRef CAS; (g) D. Reinhardt, F. Ilgen, D. Kralisch, B. Konig and G. Kreisel, Green Chem., 2008, 10, 1170–1181 RSC.
- J. P. Praly and R. U. Lemieux, Can. J. Chem., 1987, 65, 213–223 CAS.
- E. A. M. Badawi, A. S. Jones and M. Stacey, Tetrahedron, 1966, 22, 281–285 CrossRef.
- (a) M. Okahara, J. Goto and S. Komori, Kogyo Kagaku Zasshi, 1963, 66, 948–952 CAS; (b) J. G. Erickson, J. Am. Chem. Soc., 1954, 76, 3977–3978 CrossRef CAS.
- D. Plusquellec, F. Roulleau, M. Lefeuvre and E. Brown, Tetrahedron, 1990, 46, 465–474 CrossRef CAS.
- (a) D. Wells and C. J. Drummond, Langmuir, 1999, 15, 4713–4721 CrossRef CAS; (b) D. Wells, C. Fong and C. J. Drummond, J. Phys. Chem. B, 2006, 110, 12660–12665 CrossRef CAS.
- B. Helferich and W. Portz, Chem. Ber., 1953, 86, 604–612 CrossRef CAS.
- A. G. Sarap and S. P. Deshmukh, Int. J. Chem. Sci., 2009, 7, 2389-2397 Search PubMed.
- B. B. Kehm and C. W. Whitehead, Org. Synth., 1957, 37, 52.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c0gc00468e |
| This journal is © The Royal Society of Chemistry 2011 |