Mechanism of efficient anti-Markovnikov olefin hydroarylation catalyzed by homogeneous Ir(III) complexes†
Gaurav
Bhalla
a,
Steven M.
Bischof
b,
Somesh K.
Ganesh
ab,
Xiang Yang
Liu
a,
C. J.
Jones
a,
Andrey
Borzenko
a,
William J.
Tenn, III
a,
Daniel H.
Ess
bc,
Brian G.
Hashiguchi
b,
Kapil S.
Lokare
b,
Chin Hin
Leung
b,
Jonas
Oxgaard
c,
William A.
Goddard, III
*c and
Roy A.
Periana
*ab
aThe Loker Hydrocarbon Institute, Department of Chemistry, University of Southern California, Los Angeles, California 90089
bThe Scripps Energy Laboratories, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, USA. E-mail: rperiana@scripps.edu
cMaterials and Process Simulation Center, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125. E-mail: wag@wag.caltech.edu
First published on 24th November 2010
Abstract
The mechanism of the hydroarylation reaction between unactivated olefins (ethylene, propylene, and styrene) and benzene catalyzed by [(R)Ir(μ-acac-O,O,C3)-(acac-O,O)2]2 and [R-Ir(acac-O,O)2(L)] (R = acetylacetonato, CH3, CH2CH3, Ph, or CH2CH2Ph, and L = H2O or pyridine) Ir(III) complexes was studied by experimental methods. The system is selective for generating the anti-Markovnikov product of linear alkylarenes (61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :39 for benzene + propylene and 98:2 for benzene + styrene). The reaction mechanism was found to follow a rate law with first-order dependence on benzene and catalyst, but a non-linear dependence on olefin. 13C-labelling studies with CH313CH2-Ir-Py showed that reversible β-hydride elimination is facile, but unproductive, giving exclusively saturated alkylarene products. The migration of the 13C-label from the α to β-positions was found to be slower than the C–H activation of benzene (and thus formation of ethane and Ph-d5-Ir-Py). Kinetic analysis under steady state conditions gave a ratio of the rate constants for CH activation and β-hydride elimination (kCH: kβ) of ∼0.5. The comparable magnitude of these rates suggests a common rate determining transition state/intermediate, which has been shown previously with B3LYP density functional theory (DFT) calculations. Overall, the mechanism of hydroarylation proceeds through a series of pre-equilibrium dissociative steps involving rupture of the dinuclear species or the loss of L from Ph-Ir-L to the solvento, 16-electron species, Ph-Ir(acac-O,O)2-Sol (where Sol refers to coordinated solvent). This species then undergoes trans to cisisomerization of the acetylacetonato ligand to yield the pseudo octahedral species cis-Ph-Ir-Sol, which is followed by olefin insertion (the regioselective and rate determining step), and then activation of the C–H bond of an incoming benzene to generate the product and regenerate the catalyst.
:39 for benzene + propylene and 98:2 for benzene + styrene). The reaction mechanism was found to follow a rate law with first-order dependence on benzene and catalyst, but a non-linear dependence on olefin. 13C-labelling studies with CH313CH2-Ir-Py showed that reversible β-hydride elimination is facile, but unproductive, giving exclusively saturated alkylarene products. The migration of the 13C-label from the α to β-positions was found to be slower than the C–H activation of benzene (and thus formation of ethane and Ph-d5-Ir-Py). Kinetic analysis under steady state conditions gave a ratio of the rate constants for CH activation and β-hydride elimination (kCH: kβ) of ∼0.5. The comparable magnitude of these rates suggests a common rate determining transition state/intermediate, which has been shown previously with B3LYP density functional theory (DFT) calculations. Overall, the mechanism of hydroarylation proceeds through a series of pre-equilibrium dissociative steps involving rupture of the dinuclear species or the loss of L from Ph-Ir-L to the solvento, 16-electron species, Ph-Ir(acac-O,O)2-Sol (where Sol refers to coordinated solvent). This species then undergoes trans to cisisomerization of the acetylacetonato ligand to yield the pseudo octahedral species cis-Ph-Ir-Sol, which is followed by olefin insertion (the regioselective and rate determining step), and then activation of the C–H bond of an incoming benzene to generate the product and regenerate the catalyst.
Introduction
The coupling reaction of arenes and olefins to generate alkylarenes continues to be both an important C–C bond forming reaction and route to functionalize C–H bonds.1 Classically, straight-chain and branched-chain alkylbenzenes are generated by a Friedel–Crafts acylation followed by reduction or electrophilic substitution of an arene (Scheme 1a and c, respectively).2 This requires the use of halogenated hydrocarbon starting materials and several additional synthetic steps both of which generate excessive and hazardous halogenated waste. In addition, regioselectivity in these reactions is often difficult to predict and control because it is determined by the stability of the carbocation intermediate.3 Therefore, development of alternative catalysts not based on obligatory carbocation intermediates that control regio- and stereoselectivity with tolerance to a broad range of substrates and functional groups would be advantageous. This suggests that strategies based on using the C–H bond activation reaction4 followed by selective C–C bond functionalization (Scheme 1b) would minimize halogenated waste and unnecessary multistep procedures while still allowing access to these useful functionalized products. | ||
| Scheme 1 Comparison of (a) Friedel–Crafts acylation followed by Clemmenson reduction, (b) hydroarylation of olefins, and (c) Friedel–Crafts alkylation. | ||
The most practical approach to generate the formal Markovnikov (branched) hydroarylation addition was developed by Murai and co-workers using chelation-assistance to directly react olefins with acylheteroaromatics catalyzed by Ru.5 Due to the availability of a coordinating N or O, it is believed these systems operate via chelation assisted C–H activation by initial coordination of the N or O group followed by directed intramolecular C–H activation. More recently, similar O-atom chelation assistance was observed using Re, Rh, and Au complexes.6 The Goldman group has shown that Ir(PCP) pincer complexes selectively add the ortho C–H bonds of nitrobenzene and acetophenone to olefins; however this was found to be the result of thermodynamic control and not chelation assistance.7 Other groups have utilized intramolecular reactions with alkenes tethered to aryl substrates to facilitate a ring-closing hydroarylation reaction.8 Currently, Markovnikov hydroarylation catalytic systems have been limited to metals such as Pt, Pd, Ir, Au, and Ru.9
There are limitations to the route developed by Murai and Goldman because they yield only the branched (Markovnikov) products, and access to the linear (anti-Markovnikov) products are vital to a synthetic toolbox. In addition, conventional techniques (Scheme 1a or c) require the use of multi-step processes that produce large quantities of halogen-containing waste. More recently, we and others have developed a variety of systems based on Ru, Rh, Ir, and Pt that generate the preferred anti-Markovnikov selectivity in a single step reaction.10
Computational studies of such systems have revealed a delicate balance between the two steps of hydroarylation: 1) olefin insertion into the coordinated Ir-phenyl and 2) C–H activation of the arene.11 It was determined that these two steps often have an inverse relationship and the energy barrier for one step adversely affects the other. By studying several computational analogues of our bis-(acac-O,O)Ir motif and Gunnoe's Tp-Ru(Ph)(CH3CN)(CO)12 system (which reacts via the same mechanism), it was concluded that the barriers for C–H activation and olefin insertion are dependent on the energy of the metal d-orbitals. Thus, the barrier for olefin insertion increases with higher energy d-orbitals, while the barrier for C–H activation correspondingly decreases. Similar computational studies were conducted with selected systems that favor anti-Markovnikov products to reveal that both sterically bulky and electron-deficient olefins appear to favor linear products.13 Whereas more electron-rich metal/ligand systems also yield higher linear/branch ratios. In addition, the effect of sterics versus electronics on the linear to branched ratio cannot be generalized and is dependent on each specific system.
The most practical approach for the generation of anti-Markovnikov hydroarylation products was initially reported by Periana, Matsumoto, and co-workers in 2000.14 The reaction entailed C–H bond activation of benzene followed by olefin insertion to generate C–C functionalized products with selectivity towards the straight chain alkylbenzene (Scheme 1b).
The hydroarylation reaction was catalyzed by an O-donor, dinuclear, IrIII complex based on the bis-acac motif, [Acac-C–Ir]2. Throughout this paper, unless otherwise specified, acac-O,O is the O-bound, acetylacetonato or 2,4-pentanedione ligand. To further simplify discussion, the four-coordinate (acac-O,O)2IrIII motif is abbreviated as, Ir, and trans and cis refer to the orientation of the two non-spectator ligands on this motif. Thus, the abbreviation, trans-R-Ir-L, is understood to be the trans-(acac-O,O)2IrIII(L)(R) complex with the non-spectator ligands, R and L, in a trans orientation (and the two acac-O,O ligands are in a single plane). Conversely, cis-R-Ir-L is understood to be cis-(acac-O,O)2IrIII(L)(R) where the non-spectator ligands, R and L, are in a cis orientation (and the two acac-O,O ligands are not in a single plane).15,16 The related mononuclear species, (acac-O,O)2Ir(R)(L) (where R = acac, CH3, CH2CH3, Ph, CH2CH2Ph), also catalyzes rapid benzene-solvent H/D exchange. Previously reported experimental and theoretical studies revealed several important conclusions: 1) the dinuclear and mononuclear complexes follow the same mechanistic steps; 2) initiation involves either dissociation of the dinuclear complex or ligand loss from the mononuclear species to generate a reactive 5-coordinate intermediate; 3) C–H activation involves the 5-coordinate species, requiring trans to cisisomerization of the acac ligand; and 4) the C–H activation step is not the rate determining step.
Given the importance and potential broad utility of hydroarylation reactions at reducing halogenated waste and direct access to straight chain alkylbenzenes,17 we have begun a systematic study to understand the reactivity and selectivity of the bis-(acac-O,O)Ir motif in order to design more active catalysts.18 We have recently reported a bis-tropolonato IrIII analogue which shows a higher rate of C–H activation and comparable rates of hydroarylation.19 Herein, we present a detailed experimental study of hydroarylation catalyzed by O-donor IrIII complexes to expand our understanding of this system and answer the following questions:
(1) Is the observed anti-Markovnikov regioselectivity due to thermodynamic or kinetic control?
(2) Is the active catalyst a dinuclear or mononuclear species?
(3) What are the reaction orders of the substrates?
(4) Why are no β-hydride elimination products observed?
(5) What is the rate-determining step and complete mechanism for hydroarylation?
Results and discussion
The (acac-O,O)2Ir motif (Fig. 1) has been well established as both a C–H activation and hydroarylation catalyst.11,13,14,15,16 For C–H activation, the IrIII system has been shown to activate the C–H bonds of a wide variety of sp2 and sp3hydrocarbons both stoichiometrically and catalytically. The motif can tolerate a wide array of substrates and yields predominantly linear alkylarenes as a hydroarylation catalyst. Both mononuclear and dinuclear (acac-O,O)2IrIII complexes have been shown to generate a mononuclear cis-(acac-O,O)2IrIII species in situ. The most active hydroarylation catalyst previously reported was Ph-Ir–H2O, with a turnover frequency (TOF) of 1.3 × 10−2 s−1.20 Reactions of Ph-Ir–H2O in benzene with propylene and styrene led to a ratio of linear:branched products of 61:39 (benzene + propylene) and 98:2 (benzene + styrene). Due to the unique selectivity towards linear products, lack of rearrangements, and the lack of β-hydride elimination products, we were led to examine the mechanism for the hydroarylation reaction in greater detail.
 | ||
| Fig. 1 Acetylacetonato based Ir(III) O-donor complexes studied. | ||
We have previously proposed a mechanism for the hydroarylation reaction involving benzene and an olefin using the (acac-O,O)2IrIII motif as the catalyst (Scheme 2).16e The mechanism entails the dinuclear complex, [L-Ir]2 or the mononuclear complex, R-Ir-L, disassociating into a coordinatively unsaturated species to undergo a trans to cisisomerization followed by benzene C–H activation to generate a cis-iridium phenyl intermediate. The olefin then rapidly coordinates to the open coordination site to form cis-Ph-Ir-Ol (Ol = olefin). Alkene insertion into the iridium phenyl bond followed by coordination of benzene yields cis-PhCH2CH2-Ir-η2(C6H6). C–H bond activation by an oxidative hydrogen migration (OHM) mechanism releases the product and regenerates the iridium phenyl intermediate.11,13,15
![Catalytic cycle for the hydroarylation reaction using [L-Ir]2 or R-Ir-L.](/image/article/2011/GC/c0gc00330a/c0gc00330a-s2.gif) | ||
| Scheme 2 Catalytic cycle for the hydroarylation reaction using [L-Ir]2 or R-Ir-L. | ||
Catalyst stability
Initially, we examined if the reaction system was stable over reaction times >1 h, as previously reported reactions were carried out for <1 h.14,16Fig. 2 shows a plot of turnover number (TON) versus time for the hydroarylation reaction between propylene and benzene using Ph-Ir-Py.20 The linear relationship between TON and time shows that the catalyst is active and thermally stable at 180 °C over 4 h. This showcases the impressive thermal stability of the O-donor acac ligands.
Mononuclear or dinuclear catalyst
In the initial report of hydroarylation by the (acac-O,O)2Ir(III) complexes, we utilized the dinuclear complex, [Acac-C–Ir]2.14,16 Subsequently, we found that the mononuclear complexes (Ph-Ir-L, Acac-C–Ir-L where L = H2O, Py, etc.) are also active hydroarylation catalysts. While this suggests that the active catalyst is a mononuclear (acac-O,O)2Ir(R)(L) complex, it could be speculated that the active catalyst could be a dinuclear Ir species or operating via a bimolecular pathway.We have previously reported a facile conversion of these dinuclear complexes to stable mononuclear complexes when treated with coordinating ligands L (where L = pyridine),16e which is inconsistent with dinuclear complexes as the stable, active catalysts. Furthermore, study of the labile [CH3-Ir]2 complex by 1H and 13C NMR revealed that dissociation to stable 5-coordinate square pyramidal complexes (or 6-coordinate solvento complexes) occurs with an activation parameter (ΔG‡298 K) of 14.1 ± 0.5 kcal mol−1, which is lower than the ΔG‡ barrier for hydroarylation.16e
Examination of the predicted rate laws for a mononuclear catalytic species versus a dinuclear catalytic species generated in situ shows concentration dependence on L (Scheme 3). If we presume that the active catalyst is a dinuclear complex (M-M) and that it is in equilibrium with the catalyst precursor, M-L, then the rate law will be proportional to [M-L]2/[L]2 under steady state conditions. This is reasonable if the equilibrium constant for the formation of M-M or the amount of L formed from dissociation of M-L are small. Consistent with these assumptions, VT-NMR analysis of a C6D6 solution of Ph-Ir-Py at 100 °C showed no detectable free pyridine or dinuclear complexes. These results are also consistent with previous theoretical calculations that indicate that both reactions are endoergic with a ΔG > 5 kcal mol−1.11,13,15
![TOF dependence on [L] for dinuclear and mononuclear complexes.](/image/article/2011/GC/c0gc00330a/c0gc00330a-s3.gif) | ||
| Scheme 3 TOF dependence on [L] for dinuclear and mononuclear complexes. | ||
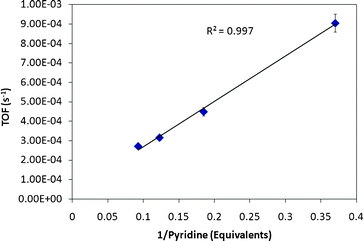
If the catalyst is the dinuclear complex M-M, plotting the TOFvs. [1/L2] should yield a straight line. On the other hand, if the active catalyst is the mononuclear species “M” generated by the loss of L from added M-L, then plotting TOFvs. [1/L] should yield a straight line at constant [M-L]. Analysis of reactions charged with varying amounts of pyridine (2–10 eq.) under standard pseudo first order conditions21 at 180 °C for 30 min in a temperature controlled oil bath, yields a linear correlation (R2 = 0.997) when 1/(equivalents of pyridine) is plotted against TOF as shown in Fig. 3. The linear correlation for Ph-Ir-Py strongly indicates a mononuclear catalyst as the active species. The system is also inhibited by pyridine suggesting a coordination catalysis mechanism.
Reaction order of substrates
Next, we wanted to examine the order of the various reactants (benzene, catalyst, and olefin) present during reaction. The reaction order on benzene was determined using Ph-Ir-Py as the catalyst in cyclohexane. Control reactions have shown that cyclohexane does not react under hydroarylation conditions and therefore can be used as an inert solvent for examining the order of benzene. A 10 mL glass Schlenk bomb flask fitted with a resealable Teflon valve and a magnetic stir bar was charged with dry, distilled benzene (0.1 to 1 mL), styrene (0.6 mL) and 0.1 mol% of catalyst from a stock solution. The solution was heated to 180 °C for 30 min in a well stirred, temperature controlled oil bath. Analysis of the reaction mixture by GC-MS and referenced to the internal standard, yields the data shown in Fig. 4. A linear relationship exists between benzene concentration and TOF, which is expected for first-order dependence on benzene. | ||
| Fig. 4 Kinetic dependence of hydroarylation on benzene concentration using Ph-Ir-Py. | ||
The hydroarylation of olefins to generate mono-alkylarenes is typically carried out at ∼200 °C with a 5–10 mM catalyst loading in 1 mL of neat arene (which acts as both reactant and solvent) and 10–20 mol% olefin. Studies of olefin concentration (with styrene as the olefin) under these conditions show a complicated dependence (Fig. 5) on concentration. The initial increase in the TOF is consistent with a first order dependence on olefin; however, higher concentrations of olefin lead to a drastic drop in catalytic rate. We previously had observed inhibition by high concentrations of olefin.15,16 Likely, high olefin concentrations leads to a ground-state stabilization effect by binding to the Ir center and thus effectively blocking arene binding that is required for catalysis.
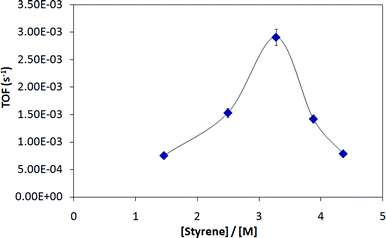 | ||
| Fig. 5 Kinetic dependence of hydroarylation on olefin concentration. | ||
As the concentration of free L during catalysis could not be easily determined, we carried out reactions in the presence of excess L (L0 > 1 eq. of added catalyst) as this allows the simplification that L = L0 if we assume that the pre-equilibrium term, K1, is small. Based on the proposed mechanism (Scheme 2)11,13,15,16 and as can be seen from the simplified rate law in Scheme 4, a first order dependence on the concentrations of catalyst, olefin and benzene is predicted along with an inverse dependence on L. We have previously shown that both C–H activation and hydroarylation show first order dependence on the catalyst (acac-O,O)2Ir(R)(L).16 As can be seen from the data there is an inverse dependence on L and a first order dependence on benzene and olefin at low pressures.
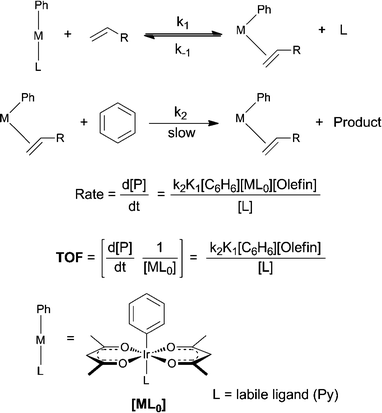 | ||
| Scheme 4 Proposed rate law for Ph-Ir-Py. | ||
Thermodynamic vs. kinetic control
A key feature of the (acac-O,O)2IrIII catalysts is the preference for anti-Markovnikov regioselectivity in olefin hydroarylation to give straight-chain (linear) alkylbenzenes. However, based on DFT calculations,11,13,15 the 40:60 ratio of iso- to n-propyl benzene can be expected based on the thermodyanmic heats of formation of the products (see Supporting Information†).22 Therefore, we wanted to examine whether the reaction is under thermodynamic or kinetic control.
For the insertion to be under thermodynamic control, it is required that a facile pathway be available for the intramolecular interconversion of the n-propyl and isopropyl benzene isomers under the conditions required for hydroarylation reaction. Importantly, such alkylarene isomerizations must not be accompanied by intermolecular trans-alkylation (reactions that transfer alkyl groups between different arenes), because in that case, the predicted thermodynamic products would be expected to be poly-alkylarenes (e.g.di-n-propylbenzene or di-iso-propylbenzene), which are not observed by GC-MS or NMR analysis. To test the possibility that a selective intramolecular alkylarene isomerization catalyst is generated, isopropyl benzene was monitored under the hydroarylation conditions (i.e., in the presence of benzene and the catalyst, Ph-Ir-Py) to detetermine if rearrangement occurs to yield the thermodynamically more stable n-propyl benzene isomer (eqn (1)). GC-MS analysis of the resulting reaction mixture showed that the isopropyl benzene remains unchanged and no n-propyl benzene was formed (Table 1). To further mimic reaction conditions, the reaction was repeated in the presence of ethylene to rule out the possibility that the potential active catalyst is generated only in the presence of olefins. Under these conditions, while ethyl benzene is observed (indicating that an active hydroarylation catalyst is present) no rearrangement of isopropyl benzene occurs and n-propyl benzene is not observed. These experiments rule out the possibility that the anti-Markovnikov selectivity stems from thermodynamic control during the hydroarylation reaction and suggest that the similarity between the thermodynamic and the observed product distribution with propylene is merely coincidental. This leads to the conclusion that the reaction operates under kinetic control.
 | (1) |
|
|
||||
|---|---|---|---|---|
| Olefin | Expected thermodynamic ratio | Observed thermodynamic ratio | ||
| Linear | Branched | Linear | Branched | |
| Propylene | 75 | 35 | 61 | 39 |
| Styrene | 97 | 3 | 98 | 2 |
| 1-Hexene | 87 | 13 | 69 | 31 |
| Isobutylene | 96 | 4 | 82 | 18 |

It is interesting to note that the same regioisomers are observed in the well documented Heck reaction.23 The common step of both the Heck and hydroarylation reactions is the insertion of the olefin into a M–Ph bond. Based on our theoretical results, electronic and steric factors both contribute to controlling the reaction selectivity.11
The olefin insertion step with the Ph–Ir olefin intermediate, cis-Ph-Ir-Ol, is proposed to control the anti-Markovnikov regioselectivity. To investigate this step, we examined the stoichiometric reaction of Ph-Ir-Py with various olefins and determined a hydrogen donor is required for the generation of alkylarenes. In the proposed catalytic reaction mechanism, this is provided by the arene co-reactant in the C–H activation step, or from the olefinvia vinylic C–H bond activation. However, since the C–H activation step is much faster than insertion, mesitylene was used as a solvent (rather than benzene) to allow for only stoichiometric reactivity. Control experiments showed that olefin hydroarylation reaction between benzene and propylene catalyzed by Ph-Ir-Py is uneffected by added mesitylene. NMR spectra of these reactions show that the organometallic complex after the reaction is either Mes-Ir-L or vinyl-Ir-L, the latter being the major product.16d,24
The stoichiometric reaction was studied using Ph-Ir-Py (15 mmol) with olefins such as propylene, ethylene, styrene and 1-hexene in liquid mesitylene at 180 °C for 20 min. The gas and liquid phases were analyzed by GC-MS to identify and quantify the reaction products. The solvent was then removed and the non-volatile reaction products were dissolved in CDCl3 and analyzed by NMR (Table 2). Addition of styrene (R = Ph) to Ph-Ir-Py in mesitylene yields 40% of the hydroarylation products, dihydrostilbene and 2,2-diphenylethane in a 98:2 ratio, along with 60% benzene (the total yield of linear + branched + benzene is 100% with respect to Ph-Ir-Py). Similarly, addition of 1-hexene (R = C4H9) leads to the formation of 1-phenyl hexane and 2-phenyl hexane in a 69:31 ratio, along with free benzene. Identical regioselectivity was observed when cis-Ph-Ir-Py was used in lieu of Ph-Ir-Py.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, where R = C6H5, CH3, or C4H9
CH2, where R = C6H5, CH3, or C4H9
|
|
||||
|---|---|---|---|---|
| R group | Linear/L | Branched (B) | L:B Ratio | Benzene (%) |
| CH3 | 40 | 27 | 61:39 |
33 |
| Ph | 39 | 1 | 98:2 |
60 |
| C4H9 | 50 | 25 | 69:31 |
25 |

In addition to hydroarylation products, a substantial amount of benzene is generated along with derivatives of the corresponding vinyl-Ir-L complex. Benzene should be the product of the microscopic reverse C–H transfer step:
Ph-Ir-Py + CH2CHR → cis-Ph-Ir-(CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CHR) + Py → cis-(CH2CR)-IR-C6H6 + Py → (CH2CR)-Ir-Py + C6H6 CHR) + Py → cis-(CH2CR)-IR-C6H6 + Py → (CH2CR)-Ir-Py + C6H6 | (2) |
During the catalytic cycle, this transformation is expected to be reversible and would yield the olefin intermediate, cis-Ph-Ir-Ol. However, in the stoichiometric reaction, the concentration of benzene is sufficiently low to trap the vinyl intermediate.
From these results, there are two important observations. First, the stoichiometric reactions of Ph-Ir-Py with olefins results in the same ratios of branched to linear alkylarene products as those observed in olefin hydroarylation catalysis. This suggests that our catalyst structure is maintained in the catalytic cycle, and no induction period is required for catalysis. Second, the presence of both the hydroarylated product andbenzene in the non-complete stoichiometric reactions shows that both C–H activation and insertion occurs in this system. However, the observed relative rates of (linear + branched) vs.benzene does not allow us to determine relative rates of insertionvs. C–H activation.
β-Hydride elimination
It is well known that metal-alkyl complexes possessing β-C–H bonds are susceptible to facile β-hydride elimination reactions.25 During hydroarylation reactions using the (acac-O,O)2Ir motif no olefin products are observed during post reaction analysis.16 Additionally, analysis of the liquid and gas phases of the stoichiometric C–H activation reactions between both PhCH2CH2-Ir-Py and CH3CH2-Ir-Py with benzene, by NMR and GC-MS showed that no olefinic products (such as styrene or ethylene) or Ir bound olefins were formed.14,16Olefin products would be expected from irreversible β-hydride elimination reactions from the coordinatively unsaturated intermediates cis-PhCH2CH2-Ir and cis-CH3CH2-Ir. The observation that only PhCH2CH2D is quantitatively formed from the stoichiometric CH activation of C6D6 with PhCH2CH2-Ir-Py is only negative evidence towards β-hydride elimination reactions occurring. Complete lack of olefinic products could suggest that β-hydride elimination reactions either do not occur or are reversible and unproductive (Scheme 5). Given these plausible possibilities, the selective formation of PhCH2CH2D from the C–H activation of C6D6 with PhCH2CH2–Ir-Py does not rule out the possibility that reversible, unproductive β-hydride elimination reactions occur. Previous DFT calculations15 have suggested that reversible β-hydride elimination is favorable, but that such reactions are reversible and unproductive. The calculated barrier for dissociative loss of olefin from the saturated, 6-coordinate hydride intermediates is higher than the barrier for the C–H activation step (Scheme 4). | ||
| Scheme 5 Reversible versus irreversible β-hydride elimination. | ||
Metal alkyls that possess β-C–H bonds, but do not react to generate olefins, could be useful in a variety of catalytic reactions (polymerization, hydroarylation, etc.). To further the understanding about the selectivity towards alkylarenes, the α-13C-labelled complex, CH313CH2-Ir-Py, was synthesized by a route analagous to CH3-Ir-Py and the C–H activation with C6D6 was examined.16d The ethyl-Ir complex was chosen because it would not show a steric or electronic bias to possible [1,2]-Ir-carbon rearrangements from reversible β-hydride elimination reactions. CH313CH2-Ir-Py was synthesized using 13C-labelled Et2Hg. As shown in Scheme 6, if reversible β-hydride elimination does occur with CH313CH2-Ir-Py, this would lead to migration of the 13C-label from the α to the β-position and formation of the 13CH3CH2-Ir-Py regioisomer. Indeed, carrying out this reaction in C6D6 led to two regioisomers of ethane: 13CH3CH2D and 13CH2DCH3 (formed by C–D activation of the C6D6 solvent and loss of 13C-ethane). The progress of the C–H activation reaction was monitored by 13C{1H} (with sufficiently long relaxation delay to afford accurate integration of the 13C resonances) and 1H NMR as shown in Fig. 6. The study of the reaction progress shows evidence for the migration of the 13C label of CH313CH2-Ir-Py (∼−9 ppm) from the α to the β-position, resulting in a formation of the β-13C regio-isotopomer, 13CH3CH2-Ir-Py (∼18 ppm).
 | (3) |
 | ||
| Fig. 6 Time dependent 13C NMR spectra for the reaction of CH313CH2-Ir-Py (−9 ppm) with C6D6 to form 13CH3CH2-Ir-Py (18 ppm) and two regioisomers of ethane (8 ppm). | ||
 | ||
| Scheme 6 Possible products expected from heating CH313CH2-Ir-Py in C6D6 to generate ethane by C–H activation showcasing reversible but unproductive β-hydride elimination. | ||
Fig. 6 also shows that a steady state concentration of the β-isomer, 13CH3CH2-Ir-Py (∼18 ppm), is generated but the concentration is substantially lower than the CH313CH2-Ir-Py present (−9 ppm). This indicates that reversible β-hydride elimination does occur and accounts for the α to β-migration of the 13C-label of CH313CH2-Ir-Py. Importantly, the lack of formation of equimolar amounts of CH313CH2-Ir-Py and 13CH3CH2-Ir-Py as ethane is lost with concomitant C–H activation of C6D6, strongly indicates that the α to β-migration of the13C-label is slower than both the C–H activation of benzene and the formation of ethane with Ph-d5-Ir-Py. This result is confirmed by analysis of the dissolved ethane produced from arene C–H activation. Thus, both regioisomers of mono-2H,13C-ethane (∼8 ppm composed of a singlet from 13CH3CH2D and a 1:1:1 triplet from 2H-13C coupling in 13CH2DCH3) are generated from the C–H activation of C6D6 as shown in eqn (2). Simulation of this pattern readily shows that the predominant ethane product is 13CH2DCH3 with ∼16 mol% of 13CH3CH2D and that this ratio is essentially constant over the course of the reaction. Analyses by 1H NMR also confirms (on the basis of the acac resonances) that Ph-d5-Ir-Py is the only new (acac-O,O)2Ir product formed after loss of ethane.
Importantly, rate measurements of the migration of the 13C-label in CH313CH2-Ir-Py and/or the relative ratio of the two regioisomers of ethane could both be expected to provide information on the relative rates of reversible β-hydride elimination versusbenzene C–H activation. This was further examined through the data gathered from NMR experiments (Fig. 6). An assumption that the formation of 13CH3CH2-Ir-Py is under steady-state conditions was applied to the kinetic analysis. It is assumed that C–H activation operates via a bimolecular pathway based on the concentration of benzene. β-hydride elimination is observed via13C migration of the ethane label suggesting a unimolecular reaction pathway. Comparison of the ratio of rate constants for the C–H activation to β-hydride elimination (kCH:kβ) reactions gives a ratio of 13CH2DCH3 to 13CH3CH2D of ∼0.5 (Scheme 7).
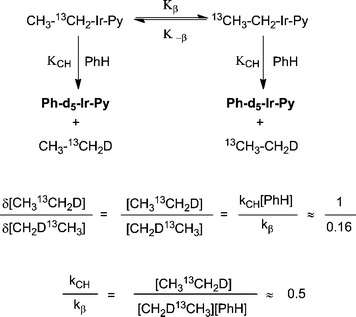 | ||
| Scheme 7 Kinetic scheme for C–H activation and α to β 13C-migration. | ||
The comparable rate constants for C–H activation and β-hydride elimination would seem to suggest a common rate determining intermediate for both processes, as was seen for the C–H activation and the trans-cisisomerization reactions of (acac-O,O)2Ir complexes.16e The intermediate is most likely, cis-R-Ir-Sol, where R = CH3CH2- in this case. Indeed, previous theoretical calculations have shown that the formation of the cis-Et-Ir-Sol intermediate is rate determining and the rates for C–H activation and hydroarylation are equivalent.11,13,15
These findings should be contrasted to the related but more electron rich IrIII complex, [Cp*(PMe3)IrR(OTf)], which undergoes irreversible β-hydride elimination to form very stable olefin metal hydrides that inhibit catalysis.26 Another closely related molecule, TpIr(olefin)2, has been shown to transform into IrIII vinyl hydride isomers, [TpIr(CHCH2)H(C2H4)], which are also the thermodynamically most stable products.27 This comparison highlights an important characteristic of the more electronegative O-donor complexes. Unlike the more electron rich compounds, the O-donor acac system avoids a thermodynamic sink of irreversible olefin binding by reducing the electron density on the Ir and allows for reversibleolefin binding.
Proposed mechanism
The proposed mechanism of the hydroarylation reaction catalyzed by the O-donor, (acac-O,O)2IrIII complexes is shown in Scheme 2. As previously reported, the trans complexes are the kinetic products that are isolated during synthesis, which upon heating lead to the quantitative formation of the thermodynamically stable cis isomers when trapped with excess L such as pyridine.16 The reaction mechanism is proposed to proceed via three key steps: 1) a pre-equilibrium, or isomerization step(s), that generates the “active catalyst”, independent of which R-Ir-L complex is used to catalyze the reaction, 2) a rate determining olefin insertion step, and 3) a C–H activation step. Unfortunately, we have been unable to synthesize or isolate the active catalytic species as they are believed to be uphill from the trans ground state and readily collapse back to the trans-R-Ir-L species upon isolation attempts.An alternative mechanism to C–H activation is the alkylation of arenes using Lewis acid catalysts. The (acac-O,O)2IrIII complexes are considered “soft” Lewis acids,1especially given the recent precedent for the use of transition metal complexes such as Au, Pt and Pd as general Lewis acids for catalyzing reactions between alkenes/alkynes and arenes.28 However, the central observation that the hydroarylation reaction catalyzed by the (acac-O,O)2IrIII complexes generate alkylarenes with anti-Markovnikov regioselectivity would tend to rule out mechanisms occurring via carbocation intermediates. Several other observations also have indicated that a carbocation intermediate is likely not involved:
1) Alkylarenes are more reactive than the parent arenes toward alkylation in Lewis acid catalyzed reactions. Alkyl benzenes are less reactive when using (acac-O,O)2Ir(R)(L) catalysts. For example, hydroarylation of ethylene with benzene shows approximately double the TON and TOF as compared to ethylbenzene.16a
2) Lewis acid catalyzed reactions are typically inhibited by 1 eq. of water. However, the (acac-O,O)2IrIII catalysts are not inhibited by the addition of several equivalents of water.16
3) Lewis acids readily catalyze the exchange of alkyl groups between alkylarenes. However, the (acac-O,O)2IrIII complexes do not catalyze reactions of diethylbenzene with benzene to generate mono-ethyl benzene under hydroarylation conditions.
4) Reaction rates of Lewis acid catalyzed reactions strongly correlate with the strength of the Lewis acid. However, while IrCl3 could be expected to be a stronger Lewis acid than the (acac-O,O)2IrIII complexes, IrCl3 does not catalyze the hydroarylation reaction under identical conditions.
5) The observation that other Ir catalysts show no hydroarylation activity or the same product selectively strongly suggests a unique catalytic activity for the (acac-O,O)2IrIII complexes.16
As can be seen in Scheme 2, it is proposed that all the various R-Ir-L as well as dinuclear ([R-Ir]2) complexes are catalyst precursors that in a series of pre-equilibrium steps (that can involve ligand loss, trans-cisisomerization, arene C–H activation and olefin coordination) lead to the same active catalyst, cis-Ph-Ir-Ol, independent of the starting complex. Mechanisms involving C–H activation16 and insertion29 are typically inner-sphere reactions and as such require a vacant coordination site on the metal for coordination of the substrate.
The ligand “L” of the O-donor (acac-O,O)2Ir(R)(L) complexes significantly influences the rate of the catalytic hydroarylation reaction. Strongly donating ligands, such as pyridine severely inhibit the catalysis; whereas, labile ligands, such as CH3OH and H2O, are weaker inhibitors. The TOFs have been correlated to the calculated relative energies of these complexes, and a linear correlation between the relative energy of the ground states and TOFs was found.15
We have previously demonstrated that when L = pyridine, exchange is facile (e.g. exchange of free pyridine with coordinated pyridine between Ph-Ir-Py and CH3-Ir-Py is rapid at room temperature and independent of the concentration of added free pyridine, as expected for a dissociative process in a pre-equilibrium step). The exchange was also examined with the cis-Ph-Ir-Py complex. Pyridine exchange with cis-Ph-Ir-Py is much slower (160 °C vs. RT) than that of its trans analog, which is consistent with theoretical predictions.11
To generate the proposed active catalyst cis-Ph-Ir-Ol, olefin coordination is required. This most likely occurs in a manner similar to conversion of trans-Ph-Ir-Py to cis-Ph-Ir-Py, i.e. by ligand dissociation, cis-transisomerization and the coordination of the olefin. However, while olefin complexes can be observed by in situNMR spectroscopy, attempts at generating and isolating such olefin complexes with various trans-R-Ir-L complexes failed, presumably due to instability. NMR analysis showed that addition of ethylene to the dinuclear complex, [Acac-Ir]2, leads to a new mononuclear complex containing coordinated ethylene. However, all attempts at isolating this complex have failed. The instability of the olefin complex(es) is consistent with the lability of the Py and H2O complexes to substitution at room temperature. Ethylene does not coordinate as strongly with (acac-O,O)2Ir(R)(L) as other π-acids (such as Py and CO), preventing isolation of an olefin complex. Furthermore, our theoretical calculations showed that the five coordinate cis-Ph-Ir species is higher in energy than the insertion transition state, suggesting that the complex undergoes further reaction under any conditions that allow for its generation.15
Since there is rapid scrambling of mixtures of benzene and deuterobenzene (relative to the hydroarylation reaction, see below), a kinetic isotope effect could not be obtained under hydroarylation conditions using mixtures of C6H6 and C6D6. However, comparison of the absolute rates of catalytic hydroarylation reactions under the same conditions (styrene and Ph-Ir-Py) in separate experiments with C6H6 and C6D6 showed no kinetic isotope effect. This result is not surprising given that olefin insertion is the rate determining step and not C–H activation as been shown previously.13 Carrying out the catalysis with ethylene and C6D6 also shows no kinetic isotope effect (KIE), and yields as the major product C6D5-CH2-CH2D (as identified by 13C NMR spectroscopy). Under these conditions, only a low level of deuterium is incorporated into unreacted ethylene as seen by GC-MS spectrometry. The possibility of a small secondary KIE is possible; however, we do not see this effect.
Comparative hydroarylation studies between trans-Ph-Ir-Py and cis-Ph-Ir-Py were carried-out at 180 °C, and it was determined that the trans isomer is 4.0 ± 0.4 times more active than the cis isomer, cis-Ph-Ir-Py. The TOF of the trans complex decreases over time, most likely because it is converted to the more thermodynamically stable cis form. However, it is possible that the active catalyst may be generated by loss of the acac ligands. Several observations indicate this is unlikely: 1) the catalyst is long lived (see Fig. 2); 2) the resting state of the catalyst after >50 turn-overs (with ethylene) is Ph-Ir-Py; 3) the O-donor (acac-O,O)2IrIII complexes are thermally stable to exchange in the presence of acac-H and protic acids such as CH3CO2H and CF3CO2H;16f 4) trans-Ph-Ir-Py is thermally stable in benzene and cleanly undergoes C–H activation and olefin insertion reactions to generate alkylarenes. Furthermore, our reported theoretical results showed that loss of an acac group costs ∼50 kcal mol−1, i.e. significantly more than the activation energy of the reaction.30 These observations strongly suggest that the (acac-O,O)2IrIII motif is part of the “active catalyst.”
Conclusion
The O-donor R-Ir-L and [R-Ir]2 complexes are active and stable catalysts for the hydroarylation of unactivated olefins and arenes. This reaction is selective to the linear alkylarenes and generates saturated products only, although it undergoes reversible and unproductive β-hydrogen elimination as determined by labeling studies. The hydroarylation reactions studied are under kinetic control. The catalyst is a mononuclear species and stable at high temperatures. A key factor is that this reaction leads in one step, directly to alkyl arenes without the production of halogentated waste while avoiding multistep procedures. Mechanistic studies suggest that the mechanism involves pre-equilibrium steps, followed by insertion of olefin, and finally C–H activation of benzene to yield the linear alkyl benzene product and regenerate the catalyst.Experimental Section
All manipulations were carried out using glovebox and high vacuum line techniques under an inert atmosphere of N2 or argon. Benzene, benzene-d6, toluene-d8 and THF were purified by vacuum transfer from sodium benzophenone ketyl. CD2Cl2 and pyridine were dried by vacuum transfer from CaH2. Synthetic work involving iridium complexes was carried out in an inert atmosphere in spite of the air stability of the complexes. Reagent-grade chemicals and solvents were used as is and purchased from Aldrich or Strem. IrCl3·H2O was purchased from Strem or Pressure Chemical. CH313CH2I was purchased from Cambridge Isotopes Inc. and was used as received. Complexes R-Ir-L, [R-Ir]2 and cis-R-Ir-L,14,16,31 and diethylmercury32 were prepared as described in the literature. Elemental analyses were done by Desert Analytics laboratory (now Columbia Analytical Services), Tucson, Arizona. Liquid phases of the reaction mixtures were analyzed with a Shimadzu GC-MS QP5000 (ver. 2) equipped with cross-linked methyl silicone gum capillary column, DB5. Gas measurements were performed using a GasPro column. The retention times of the products were confirmed by comparison to authentic samples. NMR spectra were obtained on a Bruker AC-250 (250.134 MHz for 1H and 62.902 MHz for 13C), Bruker AM-360 (360.138 MHz for 1H and 90.566 MHz for 13C), or on a Varian Mercury 400 (400.151 MHz for 1H and 100.631 MHz for 13C) spectrometer. Chemical shifts are given in ppm relative to TMS or to residual solvent proton resonances. All carbon resonances are singlets unless otherwise mentioned.Warning! Organomercury compounds are highly toxic! There is a danger of cumulative effects. These compounds may cause serious and irreversible effects on skin contact. These compounds may be fatal if absorbed through the skin - even small amounts, such as a single drop, may cause serious injury or potentially be fatal. They may cause metal fume fever if inhaled or swallowed. Chronic exposure may cause irreversible central nervous system damage, sensitization, weight loss, immunological disease and other serious effects. Work with these dangerously toxic compounds must not begin before a full assessment of the risks have been made and suitable protocols established including reading and understanding available safety information (MSDS).33
:1) as an eluent and extracted with CH2Cl2. After pumping off the solvent, the solid was redeveloped on a preparatory silica TLC plate using 1:1:2 THF–CH2Cl2:hexane. The orange band Rf = 0.87 was scraped off and extracted with CH2Cl2 and THF. The solvent was pumped off to yield an orange powder (0.0410 g, 51% yield). Characterization data matches with a previously reported sample of the non-labeled analog. 1H NMR (CD3OD): δ 5.47 (s, 2H, acac-C3H), 2.83 (dq, 1JCH = 128.5, 3JHH = 7.7, -13CH2–Ir), 1.76 (s, 12H, acac-CH3), 0.198 (m, 3H, CH3-13CH2–Ir) 13C {1H} few scans (CD3OD): δ −17.59 (s, -13CH2–Ir).
O), 149.57 (o-py), 136.32 (p-py), 124.38 (m-py), 102.76 (acac-CH), 26.66 (acac-CH3), 15.99 (d, 1JCC = 34, CH3), −10.56 (-13CH2–Ir).
Computational methodology
All calculations were performed using the B3LYP hybrid DFT functional as implemented by the Jaguar 7.0 program package.34–36 This functional has provided good agreement with experiment for (acac-O,O)2Ir(III)(R)(L) complexes and other reaction profiles of transition metal containing compounds.37,38Iridium atoms were described using the Wadt and Hay39 core-valence (relativistic) effective core potential in the LACVP basis set for geometry optimizations and single point energies with the LACVP**++ basis set.40,41 All energies reported are ΔH(298 K) = ΔE + zero point energy correction + solvation correction. Relative energies on the ΔH(298 K) surface are expected to be accurate to within 3 kcal mol−1 for stable intermediates, and within 5 kcal mol−1 for transition structures. We also note that to be consistent with past computational efforts, the methyl groups on the acac ligands were replaced with hydrogens.11Acknowledgements
We gratefully acknowledge financial support of this research by the Chevron Corporation, the University of Southern California, The Scripps Research Institute, and the Center for Catalytic Hydrocarbon Functionalization, a DOE Energy Frontier Research Center (DOE DE-SC000-1298) for financial support. We also thank Dr William Schinski for helpful discussions during the preparation of this manuscript.Notes and references
- (a) G. A. Olah, Friedel–Crafts and Related Reactions. Vol. I. General, Aspects.; Vol. II. Alkylation, and Related Reactions. Part 1.; Part 2.; 1964, 1031 pp.; 658 pp.; 703 pp. Wiley-Interscience: New York Search PubMed; (b) K. I. Goldberg and A. S. Goldman, (eds) Activation and Functionalization of C–H Bonds ACS Symp. Ser. American Chemical Society: Washington D.C. 2004, 885 Search PubMed.
- (a) M. B. Smith and J. March, March's Advanced Organic Chemistry, 6th ed.; Wiley-InterscienceNew Jersey, 2007, 719–724 Search PubMed; (b) N. Isenberg and M. Grdinic, J. Chem. Educ., 1969, 46, 601 CAS; (c) M. Grdinic and N. Isenberg, Intra-Sci. Chem. Rep., 1970, 4, 145 Search PubMed; (d) For more reviews on Friedel–Crafts chemistry since 2000, please see: M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6 DOI:10.3762/bjoc.6.6; (e) S. L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190 RSC; (f) T. B. Poulsen and K. A. Jorgensen, Chem. Rev., 2008, 108, 2903 CrossRef CAS; (g) F. Peng and Z. Shao, J. Mol. Catal. A: Chem., 2008, 285, 1 CrossRef CAS; (h) G. Busca, Chem. Rev., 2007, 107, 5366 CrossRef CAS; (i) M. Bandini, A. Melloni and A. Umani-Ronchi, Angew. Chem., Int. Ed., 2004, 43, 550 CrossRef CAS.
- G. A. Olah and Á. Molnár, Hydrocarbon Chemistry, 2nd ed.; Wiley-Interscience: New York, 2003; 229–233 Search PubMed.
- We define the C–H activation reaction to be a coordination reaction that proceeds without the involvement of free radicals, carbocations, or carbanions to generate discrete M-R intermediates (a) A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS; (b) B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154 CrossRef and citations therein; (c) R. A. Periana, G. Bhalla, W. J. Tenn, III, K. J. H. Young, X. Y. Liu, O. Mironov, C. Jones and V. R. Ziatdinov, J. Mol. Catal. A: Chem., 2004, 220, 7 CrossRef CAS and citations therein; (d) B. L. Conley, W. J. Tenn, III, K. J. H. Young, S. K. Ganesh, S. K. Meier, V. R. Ziatdinov, O. Mironov, J. Oxgaard, J. Gonzales, W. A. Goddard, III and R. A. Periana, J. Mol. Catal. A: Chem., 2006, 251, 8 CrossRef CAS; (e) R. H. Crabtree, J. Organomet. Chem., 2004, 689, 4083 CrossRef CAS; (f) M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471 CrossRef CAS.
- (a) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS; (b) S. Murai, N. Chatani and F. Kakiuchi, Pure Appl. Chem., 1997, 69, 589 CAS; (c) X. Zhang, M. Kanzelberger, T. J. Emge and A. S. Goldman, J. Am. Chem. Soc., 2004, 126, 13192 CrossRef CAS.
- (a) M. T. Wentzel, V. J. Reddy, T. K. Hyster and C. J. Douglas, Angew. Chem., Int. Ed., 2009, 48, 6121 CrossRef CAS; (b) Y. Kuninobu, T. Matsuki and K. Takai, J. Am. Chem. Soc., 2009, 131, 9914 CrossRef CAS; (c) R.-V. Nguyen, X. Yao and C.-J. Li, Org. Lett., 2006, 8, 11 CrossRef CAS.
- (a) K. Krogh-Jespersen, M. Czerw, K. Zhu, B. Singh, M. Kanzelberger, N. Darji, P. D. Achord, K. B. Renkema and A. S. Goldman, J. Am. Chem. Soc., 2002, 124, 10797 CrossRef CAS; (b) X. Zhang, M. Kanzelberger, T. J. Emge and A. S. Goldman, J. Am. Chem. Soc., 2004, 126, 13192 CrossRef CAS.
- (a) H. Harada, R. K. Thalji, R. G. Bergman and J. A. Ellman, J. Org. Chem., 2008, 73, 6772 CrossRef CAS; (b) X. Han and R. A. Widenhoefer, Org. Lett., 2006, 8, 3801 CrossRef CAS; (c) S. W. Youn, S. J. Pastine and D. Sames, Org. Lett., 2004, 6, 581 CrossRef CAS; (d) C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992 CrossRef CAS; (e) N. Marion, S. Diez-Gonzalez, P. de Fremont, A. R. Noble and S. P. Nolan, Angew. Chem., Int. Ed., 2006, 45, 3647 CrossRef CAS; (f) J. R. Chen, C. F. Li, X. L. An, J. J. Zhang, X. Y. Zhu and W. J. Xiao, Angew. Chem., Int. Ed., 2008, 47, 2489 CrossRef CAS; (g) H. H. Lu, H. Liu, W. Wu, X. F. Wang, L. Q. Lu and W. J. Xiao, Chem.–Eur. J., 2009, 15, 2742 CrossRef CAS; (h) T. S. Jian, R. Y. Tang, X. G. Zhang, X. H. Li and L. H. Li, J. Org. Chem., 2009, 74, 8834 CrossRef CAS; (i) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS; (j) C. Nevado and A. M. Echavarren, Synthesis, 2005, 167 CAS; (k) I. J. S. Fairlamb, Annu. Rep. Prog. Chem., Sect. B, 2006, 102, 50 RSC; (l) K. Komeyama, R. Igawa and K. Takaki, Chem. Commun., 2010, 46, 1748 RSC; (m) K. Xie, S. Wang, P. Li, X. Li, Z. Yang, X. An, C. C. Guo and Z. Tan, Tetrahedron Lett., 2010, 51, 4466 CrossRef CAS; (n) J. Mo and P. H. Lee, Org. Lett., 2010, 12, 2570 CrossRef CAS.
- (a) S. I. Kozhushkov, D. S. Yufit and L. Ackermann, Org. Lett., 2008, 10, 3409 CrossRef CAS; (b) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS; (c) R. A. Windenhoefer, Chem.–Eur. J., 2008, 14, 5382 CrossRef CAS; (d) L. A. Goj and T. B. Gunnoe, Curr. Org. Chem., 2005, 9, 671 CrossRef CAS; (e) K. A. Pittard, J. P. Lee, T. R. Cundari, T. B. Gunnoe and J. L. Petersen, Organometallics, 2004, 23, 5514 CrossRef CAS; (f) Y. Xiao, X. Liu and C. Che, J. Organomet. Chem., 2009, 694, 494 CrossRef CAS; (g) K. M. Gligorich, Y. Iwai, S. A. Cummings and M. S. Sigman, Tetrahedron, 2009, 65, 5074 CrossRef CAS; (h) Y. Iwai, K. M. Gligorich and M. S. Sigman, Angew. Chem., Int. Ed., 2008, 47, 3219 CrossRef CAS; (i) M. Wang, M. Wong and C. Che, Chem.–Eur. J., 2008, 14, 8353 CrossRef CAS.
- (a) R. Martinez, J. P. Genet and S. Darses, Chem. Commun., 2008, 3855 RSC; (b) Z. Zhang, X. Wang and R. A. Widenhoefer, Chem. Commun., 2006, 3717 RSC; (c) N. A. Foley, J. P. Lee, Z. Ke, T. B. Gunnoe and T. R. Cundari, Acc. Chem. Res., 2009, 42, 585 CrossRef CAS; (d) G. Bhalla, J. Oxgaard, W. A. Goddard, III and R. A. Periana, Organometallics, 2005, 24, 3229 CrossRef CAS.
- J. Oxgaard, R. A. Periana and W. A. Goddard, III, J. Am. Chem. Soc., 2004, 126, 11658 CrossRef CAS.
- (a) M. Lail, B. N. Arrowood and T. B. Gunnoe, J. Am. Chem. Soc., 2003, 125, 7506 CrossRef CAS; (b) M. Lail, C. M. Bell, D. Conner, T. R. Cundari, T. B. Gunnoe and J. L. Petersen, Organometallics, 2004, 23, 5007 CrossRef CAS; (c) N. A. Foley, M. Lail, J. P. Lee, T. B. Gunnoe, T. R. Cundari and J. L. Petersen, J. Am. Chem. Soc., 2007, 129, 6765 CrossRef CAS.
- J. Oxgaard, R. P. Muller, W. A. Goddard, III and R. A. Periana, J. Am. Chem. Soc., 2004, 126, 352 CrossRef CAS.
- T. Matsumoto, D. J. Taube, R. A. Periana, H. Taube and H. Yoshida, J. Am. Chem. Soc., 2000, 122, 7414 CrossRef CAS.
- J. Oxgaard and W. A. Goddard, III, J. Am. Chem. Soc., 2004, 126, 442 CrossRef CAS.
- (a) T. Matsumoto, R. A. Periana, D. J. Taube and H. Yoshida, J. Mol. Catal. A: Chem., 2002, 180, 1 CrossRef CAS; (b) T. Matsumoto, R. A. Periana, D. J. Taube and H. Yoshida, J. Catal., 2002, 206, 272 CrossRef CAS; (c) R. A. Periana, X. Y. Liu and G. Bhalla, Chem. Commun., 2002, 3000 RSC; (d) A. G. Wong-Foy, G. Bhalla, X. Y. Liu and R. A. Periana, J. Am. Chem. Soc., 2003, 125, 14292 CrossRef CAS; (e) G. Bhalla, X. Y. Liu, J. Oxgaard, W. A. Goddard, III and R. A. Periana, J. Am. Chem. Soc., 2005, 127, 11372 CrossRef CAS; (f) S. M. Bischof, D. H. Ess, S. K. Meier, J. Oxgaard, R. J. Nielsen, G. Bhalla, W. A. Goddard, III and R. A. Periana, Organometallics, 2010, 29, 742 CrossRef CAS.
- P. J. Chenier, Survey of Industrial Chemistry, 2nd ed.; Wiley-VCH: New York, 1992 Search PubMed.
- X. Y. Liu, W. J. Tenn, III, G. Bhalla and R. A. Periana, Organometallics, 2004, 23, 3584–3586 CrossRef CAS.
- (a) G. Bhalla and R. A. Periana, Angew. Chem., Int. Ed., 2005, 44, 1540 CrossRef CAS; (b) G. Bhalla, J. Oxgaard, W. A. Goddard, III and R. A. Periana, Organometallics, 2005, 24, 3229 CrossRef CAS.
- Turnover frequency (TOF) is defined as [(mol of product produced)/(mol of added catalysts)] per second. Turnover number (TON) is defined as [(mol of product produced)/(mol of added catalysts)] when the reaction is stopped. Moles of catalyst for the above calculations are based upon moles of added iridium.
- Reactions were run under pseudo first order conditions with the requirement that >5% conversion of starting material must occur, but no more than 10% of the starting material is converted to product.
- Chemical reaction software “Outokumpu HSC Chemistry for Windows” ver. 4, Finland.
- (a) G. T. Crisp, Chem. Soc. Rev., 1998, 27, 427 RSC; (b) W. A. Herrmann, V. P. W. Boehm and C. P. Reisinger, J. Organomet. Chem., 1999, 576, 23 CrossRef CAS.
- G. Bhalla, J. Oxgaard, W. A. Goddard, III and R. A. Periana, Organometallics, 2005, 24, 5499 CrossRef CAS.
- (a) J. P. Collman, L. S. Hegedus, J. R. Norton and R. G. Finke, Principles and Applications of Organotransition Metal Chemistry, University Science Books: Mill Valley, CA, 1987 Search PubMed; (b) J. X. McDermott, J. F. White and G. M. Whitesides, J. Am. Chem. Soc., 1976, 98, 6521 CrossRef CAS; (c) B. J. Burger, M. E. Thompson, W. D. Cotter and J. E. Bercaw, J. Am. Chem. Soc., 1990, 112, 1566 CrossRef CAS.
- (a) P. Burger and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 10462 CrossRef CAS; (b) P. O. Stoutland and R. G. Bergman, J. Am. Chem. Soc., 1988, 110, 5732 CrossRef CAS.
- C. Slugovc, I. Padilla-Martinez, S. Sirol and E. Carmona, Coord. Chem. Rev., 2001, 213, 129 CrossRef CAS.
- (a) D. Karshtedt, A. T. Bell and T. D. Tilley, Organometallics, 2004, 23, 4169 CrossRef CAS; (b) M. S. Viciu, E. D. Stevens, J. L. Petersen and S. P. Nolan, Organometallics, 2004, 23, 3752 CrossRef CAS; (c) S. W. Youn, S. J. Pastine and D. Sames, Org. Lett., 2004, 6, 581 CrossRef CAS.
- (a) S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673 CrossRef CAS; (b) M. J. Tanner, M. Brookhart and J. M. DeSimone, J. Am. Chem. Soc., 1997, 119, 7617 CrossRef CAS; (c) X. He and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 1696 CrossRef CAS.
- D. H. Ess, S. M. Bischof, J. Oxgaard, R. A. Periana and W. A. Goddard, III, Organometallics, 2008, 27, 6440 CrossRef CAS.
- M. A. Bennett and T. R. B. Mitchell, Inorg. Chem., 1976, 15, 2936 CrossRef CAS.
- H. Gilman and R. E. Brown, J. Am. Chem. Soc., 1929, 51, 928 CrossRef CAS.
- M. B. Blayney, J. S. Winn and D. W. Nierenberg, Chem. Eng. News, 1997, 75, 7 CAS Available online from the American Chemical Society at http://pubs.acs.org/cen/safety/19970512.html.
- Jaguar 7.0, Schrodinger, Inc., Portland, Oregon, 2007 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- J. Baker, M. Muir, J. Andzelm and A. Scheiner, “In Chemical Applications of Density-Functional Theory”; B. B. Laird, R. B. Ross and T. Ziegler, ed.; ACS Symposium Series 629; American Chemical Society: Washington, DC, 1996 Search PubMed.
- S. Niu and B. M. Hall, Chem. Rev., 2000, 100, 353 CrossRef CAS.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS; (b) W. A. Goddard, III, Phys. Rev., 1968, 174, 659 CrossRef; (c) C. F. Melius, B. O. Olafson and W. A. Goddard, III, Chem. Phys. Lett., 1974, 28, 457 CrossRef CAS.
- Previously, we have examined the effect of basis sets on the calculations for (acac-O,O)2Ir systems. However, it was found that the double-zeta basis set used here correlates with experiment and reduces the computational time relative to larger basis sets. Please see J. Oxgaard, R. A. Periana and W. A. Goddard, III, J. Am. Chem. Soc., 2004, 126, 11658 Search PubMed.
- (a) P. C. Hariharan and J. A. Pople, Chem. Phys. Lett., 1972, 16, 217 CrossRef CAS; (b) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Kinetic experiments. See DOI: 10.1039/c0gc00330a |
| This journal is © The Royal Society of Chemistry 2011 |