The importance of the long-chain polyunsaturated fatty acid n-6/n-3 ratio in development of non-alcoholic fatty liver associated with obesity
Rodrigo
Valenzuela
a and
Luis A.
Videla
*b
aSchool of Nutrition and Dietetics, Faculty of Medicine, Santiago-7, Chile
bMolecular and Clinical Pharmacology Program, Institute of Biomedical Sciences, Faculty of Medicine, University of Chile, Santiago-7, Chile. E-mail: lvidela@med.uchile.cl; Fax: +56-2-7372783; Tel: +56-2-9786256
First published on 19th October 2011
Abstract
Non-alcoholic fatty liver disease (NAFLD) is the most important cause of chronic liver disease that is characterized by hepatocyte triacylglycerol accumulation (steatosis), which can progress to inflammation, fibrosis, and cirrhosis (steatohepatitis). Overnutrition triggers the onset of oxidative stress in the liver due to higher availability and oxidation of fatty acids (FA), with development of hyperinsulinemia and insulin resistance (IR), and n-3 long-chain polyunsaturated FA (n-3 LCPUFA) depletion, with enhancement in the n-6/n-3 LCPUFA ratio favouring a pro-inflammatory state. These changes may lead to hepatic steatosis by different mechanisms, namely, (i) IR-dependent higher peripheral lipolysis and FA flux to the liver, (ii) n-3 LCPUFA depletion-induced changes in DNA binding activity of sterol regulatory element-binding protein 1c (SREBP-1c) and peroxisome proliferator-activated receptor α (PPAR-α) favouring lipogenesis over FA oxidation, and (iii) hyperinsulinemia-induced activation of lipogenic factor PPAR-γ. Supplementation with n-3 LCPUFA appears to reduce nutritional hepatic steatosis in adults, however, other histopathologic features of NAFLD remain to be studied.
 Rodrigo Valenzuela | Nutricionist, University of Chile (2003). Master Food Science and Nutrition (2007). PhD (c) Food Science and Nutrition. Research Scientist and Professor, Nutrition and Dietetic School, Faculty of Medicine, University of Chile, Santiago, Chile. Teaching areas: Food Science and Nutrition, Food Safety and HACCP. Research areas: Lipid in heal and disease, metabolism and cytoprotection by long-chain polyunsaturated fatty acids from marine origin, bioconversion of n-3 and n-6 fatty acids from vegetable and marine oils, physiological effects of n-3 fatty acids as functional foods. |
 Luis A. Videla | Born in Valparaíso (Chile) in February 14. Pre-graduate Studies at the University of Chile (Biochemist in 1970), Master of Sciences in Pharmacology, University of Toronto, Canada (1972), and Full Professor in 1978 (Faculty of Medicine, University of Chile). Major areas of expertise include oxidative stress mechanisms in (i) ethanol, lindane, acetaminophen, iron and copper hepatotoxicity; (ii) thyroid hormone (T3) action leading to the proposal of T3 as a liver preconditioning agent; and (iii) non-alcoholic liver disease in obese patients. At present time, alternate experimental preconditioning strategies are being assessed with the use of n-3 long-chain polyunsaturated fatty acids or iron, which may be applied in the clinical setting. |
1. Introduction
Dietary fatty acids, especially long-chain polyunsaturated fatty acids (LCPUFAs), are organic compounds essential for growth and development of mammals, including humans, as demonstrated by the pioneer work of Burr and Burr.1 This study reported that linoleic acid (C18:2 n-6, LA) and alpha-linolenic acid (C18:3 n-3, ALA) reverted an important part of noxious and deleterious effects induced by a fat-free diet in rats.1 Both AL and ALA received the designation of essential fatty acids because mammals do not have all the necessary desaturation enzymes to synthesize those fatty acids. Decades later, several investigations established the physiological importance of LA and ALA and their fatty acid derivatives in human beings, especially arachidonic acid (C20:4 n-6, ARA) for the n-6 series and eicosapentaenoic acid (C20:5 n-3, EPA) and docosahexaenoic acid (C22:6 n-3, DHA) for the n-3 series.2ARA has a significant role in developing nervous tissue, especially brain, the immune system, and vascular endothelial homeostasis, while EPA has a cardioprotective role and DHA is involved in developing nervous tissue (brain and retina)3,4 and its preservation during aging.5 The anti-inflammatory and anticoagulant properties of EPA suggested that this fatty acid could be used as part of treatments associated to chronic cardiovascular diseases.6 In this sense, EPA and DHA have been associated with multiple positive health effects,7,8 allowing the proposal of its use for the prevention of non transmissible chronic diseases,9 such as rheumatoid arthritis,10 obesity and diabetes mellitus,11 cardiovascular12 and neurodegenerative diseases,13 asthma,14 inflammatory bowel disease,8,15 cancer,16 chronic kidney failure,17 and against the injury caused to heart and liver after ischemia/reperfusion episodes.18,19 In this context, attainment of a given n-6/n-3 ratio seems to be essential for prevention and treatment of non transmissible chronic diseases, as a potential sensor for the activation of a number of mechanisms involved in inflammatory responses.20The scientific and technological development experienced during the twentieth century caused a profound impact in food production and marketing around the world, especially in the West.21 This produced significant changes in food supply and consumption,22 with one of the most significant nutritional changes in the Western diet being the drastic change in the n-6/n-3 PUFA ratio supplied by the diet. The current diet is rich in n-6 PUFA and low in n-3 PUFA, as PUFA contributors of foods are rich in n-6 PUFA, such as vegetable oils (corn, sunflower and soya) and meat (beef, chicken and pork)23,24 and poor in n-3 PUFA. Furthermore, the Western diet is characterized by low consumption of fatty fish, foods rich in n-3 PUFA, such as salmon, tuna and mackerel.23 This drastic change led to the crucial dietary imbalance in the n-6/n-3 PUFA ratio from 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 15–20:1.25 The current imbalance in the n-6/n-3 PUFA ratio is a significant change compared with the historical relationship of n-6/n-3 PUFA that humans have consumed for thousands of years,26 producing a very different situation from which man has been routinely submitted.27 A direct metabolic effect of this situation is the increase in the n-6/n-3 PUFA ratio in membrane phospholipids and derivatives of ARA, which have a coagulant and pro-inflammatory activity, resulting in an increase in cardiovascular disease mortality,28 indicating that the n-6/n-3 PUFA ratio plays a key role in health. Enhancement in the n-6/n-3 PUFA ratio may determine a pro-inflammatory state in the body favoring the development of non transmissible chronic diseases,29 especially those associated with metabolic syndrome, such as non alcoholic fatty liver disease (NAFLD) (Fig. 1).30,31 This review addresses the decrease in the n-6/n-3 PUFA ratio as a factor playing a significant role in the prevention and/or treatment of NAFLD, as enhancement in n-3 LCPUFA intake may develop an anti-inflammatory and anti-lipogenic state in the liver, decreasing the abnormal accumulation of triacylglycerols (TAGs).
:1 to 15–20:1.25 The current imbalance in the n-6/n-3 PUFA ratio is a significant change compared with the historical relationship of n-6/n-3 PUFA that humans have consumed for thousands of years,26 producing a very different situation from which man has been routinely submitted.27 A direct metabolic effect of this situation is the increase in the n-6/n-3 PUFA ratio in membrane phospholipids and derivatives of ARA, which have a coagulant and pro-inflammatory activity, resulting in an increase in cardiovascular disease mortality,28 indicating that the n-6/n-3 PUFA ratio plays a key role in health. Enhancement in the n-6/n-3 PUFA ratio may determine a pro-inflammatory state in the body favoring the development of non transmissible chronic diseases,29 especially those associated with metabolic syndrome, such as non alcoholic fatty liver disease (NAFLD) (Fig. 1).30,31 This review addresses the decrease in the n-6/n-3 PUFA ratio as a factor playing a significant role in the prevention and/or treatment of NAFLD, as enhancement in n-3 LCPUFA intake may develop an anti-inflammatory and anti-lipogenic state in the liver, decreasing the abnormal accumulation of triacylglycerols (TAGs).
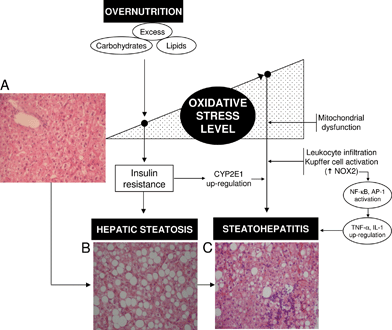 | ||
| Fig. 1 Interrelationships between the level of liver oxidative stress status and insulin resistance (IR) triggering hepatic steatosis and its progression to steatohepatitis in non-alcoholic fatty liver disease (NAFLD) associated with obesity. Overnutrition can trigger the onset of oxidative stress in the liver due to higher saturated fatty acid availability and oxidation, with consequent mitochondrial reactive oxygen species (ROS) production and antioxidant depletion. Prolonged oxidative stress might, in turn, favour n-3 long-chain polyunsaturated fatty acid depletion and IR leading to hepatic steatosis. Exacerbation of the oxidative stress status of the liver in steatohepatitis is associated with several mechanisms, including up-regulation of cytochrome P450 2E1 (CYP2E1) due to IR, mitochondrial dysfunction, and enhanced Kupffer cell and/or infiltrating leukocyte NADPH oxidase (NOX2) activity, with consequent enhancement in ROS generation. Under these conditions, promotion of hepatocellular injury might be ascribed to (i) severe oxidation of biomolecules with loss of their functions; and (ii) activation of redox-sensitive transcription factors such as nuclear factor-κB (NF-κB) and activating protein 1 (AP-1) with consequent up-regulation of the expression of pro-inflammatory mediators at the Kupffer cell level (tumor necrosis factor-α (TNF-α), interleukin (IL)-1). Hematoxylin-eosin-stained liver sections from a control patient (A) and from NAFLD obese patients with steatosis (B) or steatohepatitis (C). Magnification ×70. | ||
2. Pathogenic factors in hepatic steatosis associated with NAFLD
NAFLD is a pathological entity that is becoming a major cause of chronic liver disease associated with obesity and type 2 diabetes,32 a condition also found after the administration of amiodarone, tamoxifen, or antiretroviral drugs.33NAFLD includes the development of simple TAG accumulation in hepatocytes (hepatic steatosis) to steatosis and inflammation, fibrosis, and cirrhosis (non-alcoholic steatohepatitis, NASH) (Fig. 1). Concurrence of nutritional factors with development insulin resistance (IR) and liver oxidative stress are considered primary abnormalities leading to alterations in hepatic metabolism and the onset of steatosis (Fig. 1).31,32,34 The latter feature observed in overnutrition is associated with an increase in the peripheral mobilization of fatty acids (FAs) to the liver due to IR35,36 and enhancement in hepatic lipogenesis.37 These findings suggest that overnutrition can impose a FA overload on the liver, which is in turn associated with higher rates of mitochondrial FA oxidation and reactive oxygen species (ROS) generation,38 thus enhancing the oxidative stress status of the liver (Fig. 1).39 In agreement with these views, obese NAFLD patients with steatosis exhibit increases in oxidative stress-related parameters compared to controls, namely, (i) reduced antioxidant potential; (ii) elevated free-radical activity; (iii) enhanced Kupffer cell-dependent superoxide radical (O2˙−) formation and lipid peroxidation response; and (iv) concomitant diminution in systemic antioxidant capacity of plasma.40–48 Liver oxidative stress in obesity represents a nutritional redox imbalance resulting from a prolonged excess in oxidative load (carbohydrates and lipids) and inadequate nutrient supply (dietary antioxidants) favoring free-radical processes,49 which exhibits a causal role in multiple forms of IR.50 Exacerbation of IR and liver oxidative stress are considered crucial factors in terms of progression of steatosis to NASH in obese NAFLD patients (Fig. 1).39,483. Depletion of n-3 LCPUFA in the liver of obese NAFLD patients
Under conditions of overnutrition-induced liver oxidative stress, obese NAFLD patients exhibit two major related alterations, namely, IR and depletion of hepatic n-3 LCPUFA (Fig. 2), with concomitant enhancement in the n-6/n-3 ratio.30 Liver n-3 LCPUFA depletion in morbid obese patients with steatosis or NASH is evidenced by 50% diminution in EPA and DHA levels,30 in agreement with data in NASH patients (body mass index = 32.1 kg m−2) exhibiting n-3 LCPUFA depletion, enhanced liver lipid peroxidation, and reduced antioxidant potential over control values.51 Under these conditions, reduction in liver n-3 LCPUFA might be caused by several mechanisms. (i) Higher hepatic n-3 LCPUFA peroxidation in view of the enhanced ROS generation attained and their high susceptibility to free-radical attack with further decomposition,52 although measurement of n-3 LCPUFA-derived oxidation products in serum has not been assessed in obese patients. (ii) Defective desaturation of ALA, as suggested by the significant diminution in the product/precursor ratios (EPA + DHA)/ALA and ARA/LA pointing to Δ-5 and Δ-6 desaturase deficiency,30 which was recently confirmed by direct measurement of the activity of both liver desaturases showing 66% and 87% diminution in obese patients over control values, respectively (Fig. 2).53 Interestingly, Δ-6 desaturase inversely correlated with IR and liver oxidative stress status,53 an enzyme considered to be the rate-limiting step in PUFA synthesis that is up-regulated by insulin and down-regulated by LCPUFA.54 (iii) Dietary imbalance associated with obesity, as evidenced by a significantly lower consumption of EPA plus DHA, as well as ARA, compared to controls, concomitantly with a higher intake of transFAs (i.e., elaidic acid, 18:1, n-9trans) (Fig. 2),30 powerful Δ-6 desaturase inhibitors.55 Depletion of n-3 LCPUFA in liver phospholipids observed in obese patients also occurred in erythrocyte phospholipids, suggesting that erythrocyte FA composition could be a reliable indicator of derangements in liver lipid metabolism in obesity,56 an effect that is recovered by weight loss in association with amelioration of oxidative stress, membrane FA insaturation, and n-3 LCPUFA biosynthetic capacity.57
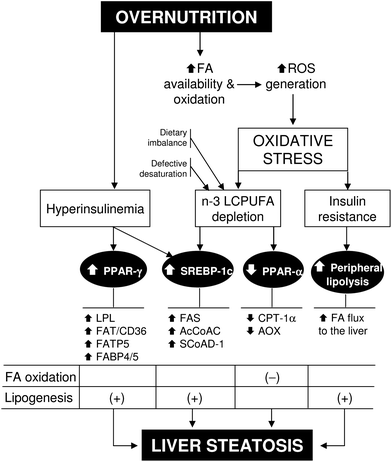 | ||
| Fig. 2 Overnutrition-induced oxidative stress and its relationship with insulin resistance, n-3 long-chain polyunsaturated fatty acid (n-3 LCPUFA) depletion, and steatosis in non-alcoholic fatty liver disease. Abbreviations: AcCoAC, acetyl-CoA carboxylase; AOX, acyl-CoA oxidase; CPT-1α, carnitine palmitoyl transferase 1-alpha; FA, fatty acid; FABP4/5, FA binding protein 4/5; FAT/CD36, FA translocase; FATP5, FA transport protein 5; FAS, FA synthase; LPL, lipoprotein lipase; PPAR-α(γ), peroxisome proliferator-activated receptor-alpha(gamma); ROS, reactive oxygen species; SCoAD-1, stearoyl-CoA desaturase-1; SREBP-1c, sterol regulatory element-binding protein 1c. | ||
N-3 LCPUFAs are important biomolecules in the regulation of hepatic lipid metabolism, which is accomplished through down-regulation of the expression and processing of transcription factor sterol regulatory element-binding protein 1c (SREBP-1c) leading to depressed lipogenic and glycogenic capacity, and up-regulation of peroxisome proliferator-activated receptor α (PPAR-α) favoring FA oxidation.58,59 Therefore, depletion of n-3 LCPUFA in the liver of obese NAFLD patients may favor FA and TAG synthesis with derangement in the capacity for FA oxidation39 and TAG export from the liver,60 leading to SREBP-1c up-regulation, PPAR-α down-regulation, and hepatic steatosis promotion (Fig. 2).61 In addition, recent studies indicate that the expression of PPAR-γ is increased in the liver of obese NAFLD patients in association with IR,62 a transcription factor favoring the expression of lipoprotein lipase, proteins involved in FA uptake, binding, and intracellular transport (Fig. 2), and liver X receptor triggering PPAR-γ expression.63,64 Considering that (i) up-regulation of PPAR-γ occurred concomitantly with that of SREBP-1c;61,62 (ii) SREBP-1c activity alone is not sufficient to account for the activation of lipogenic gene expression;65 and (iii) the lipogenic expression controlled by PPAR-γ and SREBP-1c is complementary rather than similar,63,64,66PPAR-γ up-regulation in obesity can be interpreted as a reinforcing mechanism for the lipogenic actions related to SREBP-1c induction.62
In addition to the pro-lipogenic outcome underlying liver PPAR-α down-regulation under conditions of n-3 LCPUFA depletion (Fig. 2), these changes may have pro-inflammatory implications in NAFLD patients that present substantial activation of the hepatic transcription factors nuclear factor-κB (NF-κB) and activating protein 1 (AP-1) triggering inflammation.67 This contention is based in the antagonizing action exhibited by PPAR-α on NF-κB and AP-1 activation,68 implying that down-regulation of liver PPAR-α in NAFLD may favor NF-κB and AP-1 DNA binding. In agreement with this view, significant negative correlations were established for liver NF-κB or AP-1 DNA binding and PPAR-α mRNA expression in NASH patients, with 7.8-fold and 15.1-fold increases in the respective NF-κB/PPAR-α and AP-1/PPAR-α ratios.69
4. Conclusions
The mechanisms underlying hepatic steatosis in obese NAFLD patients are multifactorial and are beginning to be understood. Overnutrition, involving excess carbohydrate and FA intake, determines an enhanced oxidative stress status in the liver due to increased FA oxidation. This redox imbalance is associated with development of IR, with secondary hyperinsulinemia, and n-3 LCPUFA depletion.30,39 Consequent steatotic mechanisms include (i) IR-dependent higher peripheral lipolysis and FA mobilization to the liver; (ii) n-3 LCPUFA depletion leading to substantial enhancement in hepatic SREBP-1c/PPAR-α ratio that favors de novolipogenesis over FA oxidation; and (iii) hyperinsulinemia-induced up-regulation of liver lipogenic factor PPAR-γ (Fig. 2). Although activation of transcription factor carbohydrate responsive element-binding protein may also play a role in obesity-related hepatic steatosis, its expression in the liver of NAFLD patients is significantly lower than in the control liver.70 In agreement with the proposed steatotic mechanisms outlined in Fig. 2, several clinical studies have reported that supplementation with fish oil, seal oil, or purified n-3 LCPUFA reduces hepatic lipid content in obese NAFLD patients, assessed either by ultrasonography71–74 or by direct measurement in post-treatment liver biopsies.75 In addition, n-3 LCPUFA administration improved circulating liver function markers,71–75 serum TAG72,73 and tumor necrosis factor-α72 levels, and hepatic microcirculatory function.71 Furthermore, a crossover randomized, controlled trial in polycystic ovarian syndrome, a condition that predisposes to hepatic steatosis, showed that fish oil diminished hepatic fat, as assessed by magnetic resonance spectroscopy.76 In conclusion, supplementation with n-3 LCPUFA significantly diminishes nutritional hepatic steatosis in adults, however, effects on histopathologic features, such as liver inflammation and fibrosis, are being addressed by clinical trials.77 Major molecular mechanisms underlying n-3 LCPUFA supplementation may involve (i) PPAR-α up-regulation and SREBP-1c down-regulation (Fig. 2) favoring FA oxidation over lipogenesis; (ii) a direct antioxidant action due to their high susceptibility to free-radical reactions,52 as shown for n-3 LCPUFA liver preconditioning against ischemia-reperfusion injury;19 (iii) up-regulation of the expression of antioxidant proteins (heme-oxygenase,78glutathione peroxidase, glutathione reductase, glutathione-S-transferases, and catalase79) or those affording glutathione repletion (glutamate cysteine ligase),78 a mechanism that may involve redox activation of NF-E2-related factor 2 (Nrf2);78 and/or (iii) down-regulation of inflammatory gene expression through interactions involving n-3 LCPUFA-dependent PPAR-α activation and further PPAR-α-mediated inhibition of NF-κB p65 sub-unit by inactive complex formation.80Acknowledgements
The studies carried out in the laboratory of the authors were funded by Grants 1060105 and 1090020 from Fondo Nacional de Desarrollo Científico y Tecnológico – FONDECYT (Chile).References
- G. O. Burr and M. M. Burr, J. Biol. Chem., 1930, 86, 587–621 CAS.
- R. T. Holman and S. B. Johnson, Am. J. Clin. Nutr., 1982, 35, 617–623 CAS.
- W. S. Harris, Curr. Atheroscler. Rep., 2009, 11, 411–417 CrossRef CAS.
- D. R. Hoffman, J. A. Boettcher and D. A. Diersen-Schade, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2009, 81, 151–158 CrossRef CAS.
- N. G. Bazan, J. Lipid Res., 2009, 50, S400–S405 CrossRef.
- S. Kanai, K. Uto, K. Honda, N. Hagiwara and H. Oda, Atherosclerosis, 2011, 215, 43–51 CrossRef CAS.
- J. W. Fetterman Jr. and M. M. Zdanowicz, Am. J. Health-Syst. Pharm., 2009, 66, 1169–1179 CrossRef.
- L. R. Ferguson, B. G. Smith and B. J. James, Food Funct., 2010, 1, 60–72 CAS.
- W. Kim, D. N. McMurray and R. S. Chapkin, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2010, 82, 155–158 CrossRef CAS.
- S. Hurst, Z. Zainal, B. Caterson, C. E. Hughes and J. L. Harwood, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2010, 82, 315–318 CrossRef CAS.
- E. Oliver, F. McGillicuddy, C. Phillips, S. Toomey and H. M. Roche, Proc. Nutr. Soc., 2010, 69, 232–243 CrossRef CAS.
- E. M. Roth and W. S. Harris, Curr. Atheroscler. Rep., 2010, 12, 66–72 CrossRef CAS.
- T. Cederholm and J. Palmblad, Curr. Opin. Clin. Nutr. Metab. Care, 2010, 13, 150–155 CrossRef CAS.
- E. Fasano, S. Serini, E. Piccioni, I. Innocenti and G. Calviello, Curr. Med. Chem., 2010, 17, 3358–3376 CAS.
- J. Bassaganya-Riera and R. Hontecillas, Curr. Opin. Clin. Nutr. Metab. Care, 2010, 13, 569–573 CrossRef CAS.
- K. M. Szymanski, D. C. Wheeler and L. A. Mucci, Am. J. Clin. Nutr., 2010, 92, 1223–1233 CrossRef CAS.
- A. N. Friedman, Seminars in Dialysis, 2010, 23, 396–400 CrossRef.
- R. Rodrigo, M. Cereceda, R. Castillo, R. Asenjo, J. Zamorano, J. Araya, R. Castillo-Koch, J. Espinoza and E. Larraín, Pharmacol. Ther., 2008, 118, 104–127 CrossRef CAS.
- J. Zúñiga, F. Venegas, M. Villarreal, D. Núñez, M. Chandía, R. Valenzuela, G. Tapia, P. Varela, L. A. Videla and V. Fernández, Free Radical Res., 2010, 44, 854–863 CrossRef.
- A. P. Simopoulos, Exp. Biol. Med., 2008, 233, 674–688 CrossRef CAS.
- J. E. Hunter, in Biological effects and nutritional essentiality, ed. C. Galli and A. P. Simopoulos, Series A: Life Sciences, Plenum Press: New York, 1989, vol. 171, pp. 43–55 Search PubMed.
- N. R. Raper, F. J. Cronin and J. Exler, J. Am. Coll. Nutr., 1992, 11, 304–308 CAS.
- A. P. Simopoulos, Prostaglandins, Leukotrienes Essent. Fatty Acids, 1999, 60, 421–429 CrossRef CAS.
- S. B. Eaton, S. B. Eaton 3rd, A. J. Sinclai, L. Cordain and N. J. Mann, World Rev. Nutr. Diet., 1998, 83, 12–23 CrossRef CAS.
- A. P. Simopoulos, Lipids, 1999, 34, S297–S301 CrossRef CAS.
- T. van Vliet and M. B. Katan, Am. J. Clin. Nutr., 1990, 51, 1–2 CAS.
- L. Cordain, B. A. Watkins, G. L. Florant, M. Kelher, L. Rogers and Y. Li, Eur. J. Clin. Nutr., 2002, 56, 181–191 CrossRef CAS.
- A. P. Simopoulos, World Rev. Nutr. Diet., 2009, 100, 1–21 CrossRef CAS.
- A. P. Simopoulos, Biomed. Pharmacother., 2006, 60, 502–507 CrossRef CAS.
- J. Araya, R. Rodrigo, L. A. Videla, L. Thielemann, M. Orellana, P. Pettinelli and J. Poniachik, Clin. Sci., 2004, 106, 635–643 CrossRef CAS.
- J. G. Gormaz, R. Rodrigo, L. A. Videla and M. Beems, Prog. Lipid Res., 2010, 49, 407–419 CrossRef CAS.
- G. C. Farrel and C. Z. Larter, Hepatology, 2006, 43, S99–S112 CrossRef.
- K. Begriche, A. Igoudjil, D. Pessayre and B. Fromenty, Mitochondrion, 2006, 6, 1–28 CrossRef CAS.
- G. Musso, R. Gambino and M. Cassader, Prog. Lipid Res., 2009, 48, 1–26 CrossRef CAS.
- S. Nielsen, Z. Guo, C. M. Johnson, D. Hensrud and M. D. Jensen, J. Clin. Invest., 2004, 113, 1582–1588 CAS.
- E. Fabbrini, B. S. Mohammed, F. Magkos, K. M. Korenblat, B. W. Patterson and S. Klein, Gastroenterology, 2008, 134, 424–431 CrossRef CAS.
- K. L. Donnelly, C. I. Smith, S. J. Schwarzenberg, J. Jessurun, M. D. Boldt and E. J. Parks, J. Clin. Invest., 2005, 115, 1343–1351 CAS.
- A. Aronis, Z. Madar and O. Tirosh, Free Radical Biol. Med., 2005, 38, 1221–1230 CrossRef CAS.
- L. A. Videla, R. Rodrigo, J. Araya and J. Poniachik, Trends Mol. Med., 2006, 12, 555–558 CrossRef CAS.
- L. A. Videla, R. Rodrigo, M. Orellana, V. Fernández, G. Tapia, L. Quiñones, N. Varela, J. Contreras, R. Lazarte, A. Csendes, J. Rojas, F. Maluenda, P. Burdiles, J. C. Díaz, G. Smok, L. Thielemann and J. Poniachik, Clin. Sci., 2004, 106, 261–268 CrossRef CAS.
- M. Orellana, R. Rodrigo, N. Varela, J. Araya, J. Poniachik, A. Csendes, G. Smok and L. A. Videla, Hepatol. Res., 2006, 34, 57–63 CrossRef CAS.
- L. Malaguarnera, R. Madeddu, E. Palio, N. Arena and M. Malaguarnera, J. Hepatol., 2005, 42, 585–591 CrossRef CAS.
- H. Cortes-Pinto, P. Filipe, M. E. Camilo, A. Baptista, A. Fernándes and M. C. de Moura, J. Português Gastroenterol., 1999, 6, 234–240 Search PubMed.
- C. P. Oliveira, J. Faintuch, A. Rascovski, C. K. Furuya Jr., S. Bastos, M. Matsuda, B. I. Della Nina, K. Yahnosi, D. S. Abdala, D. C. Vezozzo, V. A. Alves, B. ZIlberstein, A. B. Garrido Jr., A. Halpern and F. J. Carrillo, Obes. Surg., 2005, 15, 502–505 CrossRef.
- A. J. Sanyal, C. Campbell-Sargent, F. Mirshani, W. B. Rizzo, M. J. Contos, R. K. Sterling, V. A. Luketic, M. L. Shiffman and J. N. Clore, Gastroenterology, 2001, 120, 1183–1191 CrossRef CAS.
- L. Malaguarnera, M. Di Rosa, A. M. Zambito, N. dellOmbra, F. Nicoletti and M. Malaguarnera, Gut, 2006, 55, 1313–1320 CrossRef CAS.
- C. Loguercio, V. De Girolamo, I. de Sio, C. Tuccillo, F. Ascione, F. Baldi, G. Budillon, L. Cimino, A. Di Carlo, M. P. Di Marino, F. Morisco, F. Picciotto, L. Terracciano, R. Vecchione, V. Verde and C. Del Vecchio Blanco, J. Hepatol., 2001, 35, 568–574 CrossRef CAS.
- L. A. Videla, in Free radical pathology, ed. S. Alvarez and P. Evelson, Transworld Research Network, Kerala, India, 2008, pp. 369–385 Search PubMed.
- H. Sies, W. Stahl and A. Sevanian, J. Nutr., 2005, 135, 969–972 CAS.
- N. Houstis, E. D. Rosen and E. S. Lander, Nature, 2006, 440, 944–948 CrossRef CAS.
- J. P. Allard, E. Aghdassi, S. Mohammed, M. Raman, G. Avand, B. M. Arendt, P. Jalali, T. Kandasamy, N. Prayitno, M. Sherman, M. Guindi, D. W. Ma and J. E. Heathcote, J. Hepatol., 2008, 48, 300–307 CrossRef CAS.
- A. Sevanian and P. Hochstein, Annu. Rev. Nutr., 1985, 5, 365–390 CrossRef CAS.
- J. Araya, R. Rodrigo, P. Pettinelli, A. V. Araya, J. Poniachik and L. A. Videla, Obesity, 2010, 18, 1460–1463 CrossRef CAS.
- M. T. Nakamura and T. Y. Nara, Annu. Rev. Nutr., 2004, 24, 345–376 CrossRef CAS.
- U. N. Das, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2005, 72, 343–350 CrossRef CAS.
- A. Elizondo, J. Araya, R. Rodrigo, J. Poniachik, A. Csendes, F. Maluenda, J. C. Díaz, C. Signorini, C. Sgherri, M. Comporti and L. A. Videla, Obesity, 2007, 15, 24–31 CrossRef CAS.
- A. Elizondo, J. Araya, R. Rodrigo, C. Signorini, C. Sgherri, M. Comporti, J. Poniachik and L. A. Videla, Biol. Res., 2008, 41, 59–68 CrossRef.
- S. D. Clarke, Curr. Opin. Lipidol., 2004, 15, 13–18 CrossRef CAS.
- Y. B. Lombardo and A. G. Chico, J. Nutr. Biochem., 2006, 17, 1–13 CrossRef CAS.
- D. Lindén, K. Lindbeg, J. Oscarsson, C. Claesson, L. Asp, L. Li, M. Gustafsson, J. Borén and S.-O. Olofsson, J. Biol. Chem., 2002, 277, 23044–23053 CrossRef.
- P. Pettinelli, T. del Pozo, J. Araya, R. Rodrigo, A. V. Araya, G. Smok, A. Csendes, L. Gutierrez, J. Rojas, O. Korn, F. Maluenda, J. C. Diaz, G. Rencoret, I. Braghetto, J. Castillo, J. Poniachik and L. A. Videla, Biochim. Biophys. Acta, 2009, 1792, 1080–1086 CAS.
- P. Pettinelli and L. A. Videla, J. Clin. Endocrinol. Metab., 2011, 96, 1424–1430 CrossRef CAS.
- U. A. Boelsterli and M. Bedoucha, Biochem. Pharmacol., 2001, 63, 1–10 CrossRef.
- S. J. Bensinger and P. Tontonoz, Nature, 2008, 454, 470–477 CrossRef CAS.
- G. Liang, J. Yang, J. D. Horton, R. E. Hammer, J. L. Goldstein and M. S. Brown, J. Biol. Chem., 2002, 277, 9520–9528 CrossRef CAS.
- J. D. Horton, J. L. Goldstein and M. S. Brown, J. Clin. Invest., 2002, 109, 1125–1131 CAS.
- L. A. Videla, G. Tapia, R. Rodrigo, P. Pettinelli, D. Haim, C. Santibañez, A. V. Araya, G. Smok, A. Csendes, L. Gutierrez, J. Rojas, J. Castillo, O. Korn, F. Maluenda, J. C. Díaz, G. Rencoret and J. Poniachik, Obesity, 2009, 17, 973–979 CrossRef CAS.
- J. George and C. Liddle, Mol. Pharmaceutics, 2008, 5, 49–59 CrossRef CAS.
- L. A. Videla, Clin. Nutr., 2010, 29, 687–688 CrossRef.
- N. Higuchi, M. Kato, Y. Shundo, H. Tajiri, M. Tanaka, N. Yamashita, M. Kohjima, K. Kotoh, M. Nakamuta, R. Takayanagi and M. Enjoji, Hepatol. Res., 2008, 38, 1122–1129 CrossRef CAS.
- M. Capanni, F. Calella, M. R. Biagini, S. Genise, L. Raimondi, G. Bedogni, G. Svegliati-Baroni, F. Sofi, S. Milani, R. Abbate, C. Surrenti and A. Casini, Aliment. Pharmacol. Ther., 2006, 23, 1143–1151 CrossRef CAS.
- L. Spadaro, O. Magliocco, D. Spampinato, S. Piro, C. Oliveri, C. Alagona, G. Papa, A. M. Rabuazzo and F. Purrello, Dig. Liver Dis., 2008, 40, 194–199 CrossRef CAS.
- F. S. Zhu, S. Liu, X. M. Chen, Z. G. Huang and D. W. Zhang, World J. Gastroenterol., 2008, 14, 6395–6400 CrossRef CAS.
- A. Hatzitolios, C. Savipoulos, G. Lazaraki, I. Sidiropoulos, P. Haritanti, A. Lefkopoulos, G. Karagiannopoulou, V. Tzioufa and K. Dimitrios, Indian J. Gastroenterol., 2004, 23, 131–134 Search PubMed.
- N. Tanaka, K. Sano, A. Horiuchi, E. Tanaka, K. Kiyosawa and T. Aoyama, J. Clin. Gastroenterol., 2008, 42, 413–418 CrossRef CAS.
- A. J. Cussons, G. F. Watts, T. A. Mori and B. G. A. Stuckey, J. Clin. Endocrinol. Metab., 2009, 94, 3842–3848 CrossRef CAS.
- H. Shapiro, M. Tehilla, J. Attal-Singer, R. Bruck, R. Luzzatti and P. Singer, Clin. Nutr., 2011, 30, 6–19 CrossRef CAS.
- L. Gao, J. Wang, K. R. Sekhar, H. Yin, N. F. Yared, S. N. Schneider, S. Sasi, T. P. Dalton, M. E. Anderson, J. Y. Chan and J. D. Morrow, J. Biol. Chem., 2007, 282, 2529–2537 CrossRef CAS.
- A. Demoz, N. Willumsen and R. K. Berge, Lipids, 1992, 27, 968–971 CrossRef CAS.
- D. B. Jump, Curr. Opin. Lipidol., 2008, 19, 242–247 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |