Mechanisms of action of isothiocyanates in cancer chemoprevention: an update†
Sandi L.
Navarro
ab,
Fei
Li
a and
Johanna W.
Lampe
*ab
aFred Hutchinson Cancer Research Center, Division of Public Health Sciences, 1100 Fairview Ave., N M4-B402, Seattle, WA 98109, USA. E-mail: jlampe@fhcrc.org; Fax: +1 (206) 667 7850; Tel: +1 (206) 667 6580
bInterdisciplinary Graduate Program in Nutritional Sciences, Department of Epidemiology, University of Washington, Seattle, WA 98195, USA
First published on 21st September 2011
Abstract
Isothiocyanates (ITC), derived from glucosinolates, are thought to be responsible for the chemoprotective actions conferred by higher cruciferous vegetable intake. Evidence suggests that isothiocyanates exert their effects through a variety of distinct but interconnected signaling pathways important for inhibiting carcinogenesis, including those involved in detoxification, inflammation, apoptosis, and cell cycle and epigenetic regulation, among others. This article provides an update on the latest research on isothiocyanates and these mechanisms, and points out remaining gaps in our understanding of these events. Given the variety of ITC produced from glucosinolates, and the diverse pathways on which these compounds act, a systems biology approach, in vivo, may help to better characterize their integrated role in cancer prevention. In addition, the effects of dose, duration of exposure, and specificity of different ITC should be considered.
 Sandi L. Navarro | Sandi L. Navarro received her PhD in August 2011, with a major in nutritional sciences and a minor in public health genetics from the University of Washington in Seattle. Her dissertation research focused on the effects of cruciferous vegetables on biomarkers of systemic inflammation, and modulation of response by genotype, using a controlled feeding trial in humans. After graduation, she began post-doctoral training at the Fred Hutchinson Cancer Research Center in the area of metabolomics and systems biology approaches to characterizing effects of dietary interventions. |
 Fei Li | Fei Li received his PhD degree in nutritional sciences from the University of Washington in 2010. Since then he has conducted research as a post-doctoral fellow at Fred Hutchinson Cancer Research Center. His dissertation research examined the association between cruciferous vegetable consumption and human gut microbiota structure, and the gut bacterial metabolism of bioactive cruciferous vegetable constituents. His current research is focused on the relationship between dietary components, human gut microbiota and risk factors for cancer and other diseases. |
 Johanna W. Lampe | Johanna W. Lampe is a Full Member in the Public Health Sciences Division at Fred Hutchinson Cancer Research Center and a Research Professor in the Department of Epidemiology at the University of Washington in Seattle. Dr. Lampe earned a PhD in nutritional sciences, with a minor in biochemistry, at the University of Minnesota. Her research program addresses the effects of plant-food constituents on cancer susceptibility in humans using controlled dietary interventions. Her lab also studies the interindividual variation in gut bacterial metabolism of phytochemicals and the impact of diet on the gut microbiome in relation to human health. |
Introduction
Higher consumption of cruciferous vegetables (from the Brassicaceae plant family; e.g., broccoli, cabbage, Brussels sprouts, watercress, kale, cauliflower) is associated with a reduced risk of several cancers, particularly cancers of the gastrointestinal tract, lung and prostate.1–4 Similarly to many other plant foods, crucifers contain various compounds associated with reduced cancer risk including fiber, carotenoids, lutein, flavonoids, phytosterols, folic acid and vitamin C.5 In contrast to other plants however, cruciferous vegetables also contain substantial amounts of sulfur-containing glucosinolates, which, on hydrolysis by the enzymes myrosinase in the crucifers or β-thioglucosidases in certain gut bacteria, are converted to biologically active compounds such as indoles and isothiocyanates (ITC), and less active nitriles.6 These bioactive compounds are hypothesized to be responsible for the chemoprotective effects conferred by cruciferous vegetable consumption above and beyond the protective effects of higher intake of fruits and vegetables in general.Glucosinolate profiles
Glucosinolates are a class of sulfur-rich secondary plant metabolites. Although derived from the same botanical family, the glucosinolate composition of different cruciferous vegetables varies substantially (Fig. 1).7–10 For example, broccoli is a rich source of the glucosinolate glucoraphanin, cabbage is rich in sinigrin, and watercress is high in gluconasturtiin. The type and amount of glucosinolates produced by the plants depend on environmental factors such as temperature, hydration, presence of iron, insects, and soil pH (sulfur and nitrogen content), although the ratio of individual glucosinolates remains relatively constant within each specific plant.2 There are over 150 known glucosinolates,2 which all share a common sulfur-linked β-D-glucopyranose structure, but differ in side chains. These side chains are derived from different amino acids during glucosinolate synthesis in plant cells. The glucosinolates can be divided into several sub-groups based on the chemical structure of the side chains (Fig. 1).8,11,12 For example, the alkylthioalkyl side-chain of glucoraphanin contains a sulfur group, whereas, the aromatic side-chain of gluconasturtiin contains a phenethyl group.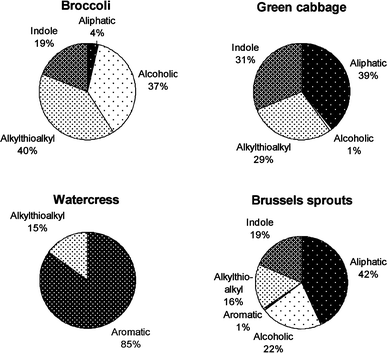 | ||
| Fig. 1 Glucosinolates in cruciferous vegetables can be divided into several sub-groups based on the chemical structure of the side chains. | ||
Following metabolism of glucosinolates into ITCs in vivo, the structural difference of the glucosinolate is conferred to that of the cognate ITC [e.g., glucoraphanin to sulforaphane (SFN), sinigrin to allyl ITC (AITC), and gluconasturtiin to phenethyl ITC (PEITC) Fig. 2]. The biological effect of ITCs varies due to the side-chain structure. For instance, in vitro studies have shown that SFN is taken into cells faster, kept intracellularly longer, and at higher accumulations than several other ITCs,13,14 and has the highest potency of inducing the expression of two phase 2 enzymes: glutathione S-transferase (GST) and quinone reductase (QR).13,15 In contrast, Jakubikova et al.16 showed that AITC was most effective in causing HL60 cell cycle arrest, while PEITC and benzyl ITC (BITC)17 were the most effective in inducing apoptosis, among six different ITCs. Prawan, et al.18 studied the effect of ten synthetic ITC analogs on pro-inflammatory NF-κB activity in vitro, and reported that subtle changes in ITC structure had a profound impact on inhibition potential. Therefore, in addition to the amount consumed, the variety of cruciferous vegetables ingested may also influence biologic response. To date, differential effects of cruciferous vegetables with diverse glucosinolate profiles have not been directly compared in vivo in humans.
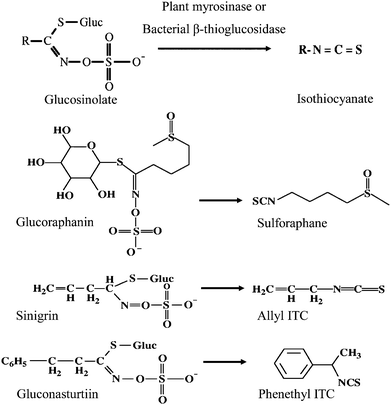 | ||
| Fig. 2 Conversion of select glucosinolates to their corresponding isothiocyanates. | ||
Mechanisms of action
Enthusiasm for the study of the chemoprotective effects of ITCs was initially generated by early work in animal models demonstrating inhibition of carcinogen-induced tumor formation at various sites with administration of ITCs.19–23 These studies supported the epidemiologic literature suggesting a cancer protective effect of cruciferous vegetable consumption. Subsequently, evidence has emerged to suggest that ITCs may be involved in a number of other distinct but interconnected signaling pathways important for inhibiting carcinogenesis, including those involved in detoxification, inflammation, apoptosis, and cell cycle and epigenetic regulation, among others (Fig. 3). In addition to ITCs, other glucosinolate hydrolysis products include indoles and nitriles, which have also been purported to have chemoprotective activity. An in-depth discussion of other glucosinolate products is beyond the scope of the present review and the reader is referred to other work on the topic.24–27 Numerous comprehensive reviews have been published on the biologic activities of ITCs over the past several years, however, this field continues to evolve and remains an active area of research.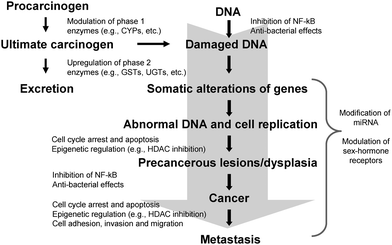 | ||
| Fig. 3 Mechanisms of action of isothiocyanates in the modulation of signaling pathways involved in cancer chemoprevention. (CYP = cytochrome P450; GST = glutathione S-transferase; UGT = UDP-glucuronosyl transferase; NF-κB = nuclear factor kappa B; HDAC = histone deacetylase; miRNA = micro RNA). | ||
Modulation of phase 1 and phase 2 biotransformation enzymes
In studies designed to elucidate the mechanisms of cancer prevention by ITCs, Wattenberg21,28 found that ITC administration increased carcinogen metabolism and detoxification, resulting in decreased initiation and promotion of tumors in carcinogen-challenged rats. Based on this seminal work, it was hypothesized that the major mechanism of chemoprevention by ITCs was through modulation of biotransformation enzymes [phase 1, e.g., cytochrome P450 (CYP) family, and phase 2, e.g., glutathione S-transferases (GST), UDP-glucuronosyl transferases (UGT), etc.].29,30 GST and UGT are multigene families of enzymes involved in conjugation of exogenous compounds (e.g., various carcinogens and other xenobiotics) and endogenous compounds (e.g., sex steroid hormones) implicated in cancer risk.31Most in vitro evidence suggests that up-regulation of phase 2 biotransformation enzymes by ITCs occurs through interaction of ITCs with the cytoplasmic-anchoring protein Kelch-like ECH-associated protein 1 (Keap 1), which represses the transcription factor NF-E2-related factor 2 (Nrf2).32,33 ITC interaction with critical cysteine residues of Keap1 results in dissociation of Keap1 from Nrf2, allowing Nrf2 translocation to the nucleus and gene activation via the antioxidant response element (ARE) located upstream of the promoter region of the genes of many antioxidant and phase 2 biotransformation enzymes,32,33 including GST,34 UGT,35 and NAD(P)H:quinone oxidoreductase-1 (NQO1),36 among others. Participation of Nrf2 in regulation of genes involved in carcinogen metabolism and antioxidant activity has been extensively reviewed.37–41
The effects of ITC-mediated upregulation of phase 2 enzymes have been corroborated in feeding studies in humans. For example, we showed previously that consumption of cruciferous vegetables for two weeks compared to a diet devoid of fruits and vegetables increased GST-α concentrations, and decreased serum bilirubin concentrations, indicative of increased UGT1A1 activity.42,43 Gasper and colleagues44 observed increased expression of several xenobiotic metabolizing enzymes in gastric mucosa after individuals consumed high-glucosinolate broccoli.
There is also some evidence to suggest that ITCs directly inhibit phase 1 enzymes, although to a lesser extent, via both competitive inhibition as well as direct covalent modification (reviewed in22,45). Phase 1 enzymes are involved in metabolic activation of endogenous and exogenous compounds, including potential carcinogens. However, in addition to ITCs, glucosinolate hydrolysis leads to the production of indoles and nitriles. Indoles induce both phase 1 and 2 enzymes through binding with the aryl-hydrocarbon receptor (AhR) and subsequent interaction with the xenobiotic response element (XRE).27,46,47 Crambene, a nitrile, has been shown to act similarly to ITCs and interact with the ARE.27 Generally, induction of both phase 1 and phase 2 enzymes is thought to accelerate metabolism of carcinogens toward elimination.
As cruciferous vegetables contain a mixture of ITCs, indoles and nitriles, consumption of the whole food, versus single, isolated compounds, may confer protection beyond that of individual compounds. Thus, translation of studies based on isolated compounds to intake data in humans is very complex, and points to the need for further in vivo studies evaluating the effects of these vegetables on biologic outcomes in humans. The impact of ITCs on biotransformation has been extensively reviewed.22,24,45,48–52
Inhibition of nuclear factor kappa B (NF-κB) pathways
Further study on the chemopreventive potential of ITCs led to the discovery that ITCs may play a role in several other signaling pathways critical to carcinogenesis, including inhibition of NF-κB.24 NF-κB is a transcription factor and central mediator in the activation of genes involved in the inflammatory process, e.g., cytokines, chemokines, adhesion molecules and other soluble factors involved in the immune response.53 While acute inflammation is beneficial in some instances (e.g., injury or infection), persistent low-level inflammation is associated with predisposition to several chronic diseases.54–57 ITCs have been shown to inhibit NF-κB-mediated processes in vitro24,50,58–60 and in vivo, in animal models,61,62 and may therefore reduce inflammation—a well-recognized risk factor in carcinogenesis.63 For instance, constitutive activation of NF-κB is common in colon, liver and prostate cancer, and leads to upregulation of a number of cytokines, growth factors and anti-apoptotic genes.50,64,65 ITC-mediated inhibition of NF-κB-regulated pathways is mentioned in several reviews,24,50,66,67 however, a dedicated work on the subject is still lacking.The NF-κB family of transcription factors is composed of heterodimeric complexes of proteins from the Rel family, the most abundant of which is the p65/p50 heterodimer.61 NF-κB is retained in an inactive form in the cytoplasm by an inhibitor molecule, inhibitor kappa B (IκB).68 Following a pro-inflammatory stimulus or oxidative event, IκB is phosphorylated via the IκB kinase complex (IKK) and ubiquitinated resulting in degradation and subsequent liberation of the NF-κB complex. ITC-mediated inhibition of NF-κB has been attributed to repression of IKK phosphorylation, preventing IκB degradation and thereby inhibiting transcriptional induction by NF-κB.59 This inhibition results in decreased expression of numerous NF-κB target genes, most notably inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), interleukin 6 (IL-6), and tumor necrosis factor alpha (TNF-α).50,59,69 There is also some evidence that SFN binds directly to essential thiol groups of p50, a functionally active subunit of NF-κB, thereby inhibiting NF-κB-binding to DNA.70
In addition to direct inhibition of NF-κB activity, it has been proposed that ITC-activation of Nrf2 may also lead to inhibition of NF-κB indirectly via crosstalk between Nrf2 and NF-κB transcription factors.71–73 This hypothesis is based on several observations. Nair et al.74 proposed a regulatory network for coordinated modulation of Nrf2 and NF-κB through a common mitogen-activated protein kinase (MAPK) cascade, based on common regulatory sequences in the transactivation domains of Nrf2 and NF-κB, and key regulatory genes in inflammatory signatures. Additionally, Khor et al.75 reported decreased levels of anti-oxidant/phase 2 enzymes with simultaneous up-regulation of the NF-κB-regulated pro-inflammatory mediators COX-2, iNOS, IL-1, IL-6, and TNF-α in Nrf2 knock-out mice with chemically-induced colitis. Ma et al.76 also observed a lupus-like autoimmune syndrome in Nrf2-deficient mice, marked by multi-organ inflammatory lesions and premature death due to rapid progression of nephritis. Taken together, these data suggest that Nrf2 is involved not only in the activation of antioxidant/phase 2 gene transcription machinery, but also in the suppression of pro-inflammatory signaling.
This finding is consistent with previous observations of an anti-inflammatory role for Nrf2.71,77 Although the mechanism for this inhibition has yet to be worked out, direct evidence of the reverse (i.e., inhibition of Nrf2 by NF-κB) has been well-documented. Liu et al.78 demonstrated antagonism of Nrf2 by NF-κB at the transcriptional level through competition for co-activator CREB-binding protein (CBP) required for translocation, and concomitant recruitment of histone deacetylase (HDAC), a co-repressor. The potential cross-talk between NF-κB and Nrf2 requires further study, but may explain part of the cancer-protective effects of ITCs. Thus, Nrf2 may be a key mediator through which both phase 2 biotransformation and inflammation pathways are modulated.
Epigenetic regulation
Epigenetic regulation is a modification of DNA without a change in the sequence, that results in a change in gene expression or phenotype. Unlike mutations in the genetic code, these epigenetic alterations, e.g., histone modification and methylation, may be modifiable. Recent evidence suggests that constituents in the diet, including ITCs, have the potential to alter a number of these epigenetic events (reviewed in ref. 24, 50, 79–82).One form of epigenetic regulation is the modification of histone proteins. When histones are acetylated, DNA is accessible to transcription factors. Conversely, when acetyl groups are removed from histones through the action of histone deacetylases (HDAC), access to DNA by transcription factors is restricted, and transcription is suppressed. As such, coordination of histone acetylation and deacetylation is an important regulatory mechanism for gene expression. In cancer, the balance between acetylation and deacetylation is often dysregulated, and tumor suppressor genes are frequently silenced.83 Inhibition of HDAC has been shown for ITCs both in vitro and in vivo in animal models and may alter tumorigenesis.84–88 Associations between inhibition of HDAC activity and increases in gene expression have been demonstrated with the tumor suppressor gene p2189,90 and the pro-apoptotic gene Bax.84 Interestingly, it appears that SFN metabolites (sulforaphane-cysteine and sulforaphane-N-acetyl cysteine) rather than the parent compound may be responsible for the inhibition, possibly by acting as competitive inhibitors.86
ITCs can also exert an epigenetic effect via modulation of DNA methylation. Meeran et al.91 showed that DNA methyltransferases were down-regulated by SFN in breast cancer cells, which led to site-specific CpG island demethylation in the telomerase reverse transcriptase gene. Another study by Wang et al.92 showed a CpG island demethylation effect of PEITC on the GSTP1 gene in prostate cancer cells, resulting in a significant increase in enzyme expression and activity. Lu et al.88 examined the effect of phenylhexyl ITC on myeloma cells. This ITC not only inhibited HDAC, but also induced DNA demethylation of the p16 gene, which led to reactivation of this tumor suppressor gene in myeloma cells. While potential modification of epigenetic events may be a significant chemoprotective property, how ITCs are interacting in these pathways is, thus far, poorly understood. This is currently an active area of research however, and we can anticipate further characterization of these interactions in the near future.
Micro RNA (miRNA) regulation
miRNAs are a recently discovered class of post-transcriptional regulators that bind to target messenger RNA transcripts, usually resulting in gene silencing.93 This is an exciting new area of research, particularly in the area of cancer prevention. Two recently published studies in animal models suggest that ITCs may have the potential to modulate miRNA regulation.94,95 Using microarray and quantitative PCR methods, investigators found that the altered expression of a number of miRNA molecules, triggered by environmental cigarette smoke, can be attenuated by PEITC.94,95 A variety of these small noncoding RNA molecules are known to have regulatory functions in cell proliferation, differentiation and apoptosis, Ras activation, p53 signaling, NF-κB inhibition, etc. Therefore, modulation of miRNA expression by ITCs may be another avenue through which ITCs exert a chemoprotective effect.Stimulation of cell cycle arrest and apoptosis
Suppression of tumor cell growth in culture and animal models has been reported with several ITCs, and, while the molecular processes of this suppression are still uncertain, investigations have provided some potential ITC targets. In prostate cancer cells, treatment with either SFN or PEITC resulted in cell cycle arrest in the G2/M phase of the cell cycle and concomitant decrease in concentrations of a number of cell division cycle (Cdc) regulators (e.g., Cyclin B1, Cdc25B and Cdc25C), required for progression into M-phase.96,97 Similarly, AITC has been shown to inhibit survival of brain malignant glioma GBM 8401 cells in a dose-dependent manner via G2/M arrest with parallel reduction in cyclin dependent kinase 1 (CDK1)/Cyclin B activity.98 Mi et al.99 reported inhibition of proteosomal protein degradation and dysruption of microtubule formation in multiple myeloma cells. In vivo, mice with mutations in one allele of the APC gene (adenomatous polyposis coli; APCnull/+) treated with 300 or 600 ppm SFN in their diet for three weeks developed fewer and smaller intestinal adenomas compared to mice on a control diet.100 The investigators also reported increased apoptotic activity and lower cell proliferation in mice fed the ITC-containing diet.Apoptosis can be achieved through mitochondrial/intrinsic pathways, death-receptor cascades/extrinsic pathways, which converge on down-stream effector caspase-3, or caspase-independent pathways.51In vitro studies have reported apoptosis in response to treatment with ITCs through a number of targets at different points in these pathways. These include down-regulation of anti-apoptotic Bcl-2 and up-regulation of pro-apoptotic Bax expression,101 proteolytic activation of caspase-3,101 decrease in mitochondrial potential with subsequent release of apoptotic generating proteins cytochrome c and inhibitor of apoptosis (IAP) family member Smac/DIABLO,102 activation of parallel MAPK cascades, (e.g., extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38),51 as well as others. It is not entirely clear how ITCs initiate intracellular signaling leading to apoptosis. One mechanism put forth to explain these observations is that administration of ITCs to cells in high concentrations results in the generation of reactive oxygen species (ROS) and consequent depletion of intracellular glutathione.24 Alternatively, Xiao, et al.103 proposed ITC-inhibition of oxidative phosphorylation as a means of ROS production, based on observations of PEITC interaction with mitochondrial complex III in prostate cancer cells. Mi and colleagues99 recently reported ITC-induced apoptosis through inhibition of proteasome activity with marked accumulation of the tumor suppressor p53 in multiple myeloma cells, independent of ROS generation. These results indicate that ITCs likely elicit apoptosis through a variety of pathways. Moreover, there is evidence to suggest that the cytotoxic effects of ITCs may selectively target cancer, rather than normal cell types.104 Numerous papers exist on the molecular basis of cell cycle regulation and induction of apoptosis.51,67,105–108
Modulation of hormone receptor expression
The complexing of androgen or estrogen with their receptors can activate transcription of numerous genes involved in cell proliferation. Certain types of cancers, such as breast and prostate, have been linked to dysregulation of sex hormone-mediated gene expression.109,110 Although modulation of sex steroid metabolism by crucifers, mainly indoles, has been documented,26,111 few studies have investigated the effects of ITCs on the expression of sex hormones and their receptors. In an in vitro prostatic cell study, Beklemisheva et al.112 showed that PEITC suppressed testosterone-induced cell growth by down-regulating Sp1-mediated androgen receptor transcription. More recent work by Kang et al.113 found that ITCs can repress the expression of estrogen receptor (ER)-α in human breast cancer cells. These investigators found that the expression of an estrogen responsive gene, pS2, was significantly reduced after ITC treatment due to abrogation of estrogen and receptor interaction.113 Ramirez et al.114 reported that progesterone receptor expression, in addition to ER-α expression, was also inhibited by SFN. Interestingly, Telang et al.115 demonstrated up-regulation of the ER-β gene in human mammary cells by both SFN and PEITC, indicating that ITCs may serve as a modulator for the ratio of ER-α- to -β-subtype concentrations. ER subtypes have been shown to have opposing actions on AP1-dependent genes, including those involved in mammary proliferation and cell growth.116 ER-α generally enhances proliferation whereas ER-β has been shown to reduce proliferation by stimulating cell cycle arrest in the G2.115 In the work by Telang et al.,115 changes in the ER subtype ratio paralleled increases in the pro-apoptotic gene BAD, and tumor suppresser genes, p21 and p27.115Anti-angiogenic and anti-metastatic effects
Angiogenesis and metastasis are key steps in the development of malignant cancer. The growth of new blood vessels is essential for the formation of large tumors because of the high demand for oxygen and nutrient import, and the necessity of waste export for the fast-growing tumor cells. The anti-angiogenic effects of ITCs have been reviewed recently by Cavell et al.117 Studies have shown that ITCs modulate tumor angiogenesis via several different pathways. ITCs can down-regulate the expression of pro-angiogenic genes such as vascular endothelial growth factor (VEGF) by targeting hypoxia inducible factor (HIF) transcription factors. ITCs can also inhibit other factors involved in tumor angiogenesis such as NF-κB, activator protein-1 (AP1) and MYC, independent of the HIF pathway. Tubulin, a protein required for cell morphogenesis and migration during the tumor angiogenesis process, has also been recognized as a target for ITCs. ITCs not only inhibit tubulin polymerization118,119 but also promote tubulin degradation in cancer cells.120,121Metastasis, or the spread of tumor cells, both locally and to distant places via lymphatic or blood vessels, is another indication of cancer progression. ITCs and derivatives were shown to inhibit cell adhesion, invasion and migration in vitro, and suppress metastasis in vivo through down-regulation of matrix metalloproteinase (MMP) and up-regulation of tissue inhibitors of matrix metalloproteinase (TIMP). Several metastatic biomarkers were suppressed after SFN treatment in animal models.122,123 Epidemiologic studies also indicate that crucifer intake is inversely associated with survival of many cancers.124–126
Anti-bacterial effects
From the point of view of the plant, ITCs act as a deterrent for insects, suppress the growth of other nearby plants and provide antibiotic properties, warding off invading pathogens.2 The antibacterial effects of ITCs in humans were first described by Fahey et al.127 with Helicobacter pylori (H. pylori). SFN was shown to inhibit the growth of this bacterium, which is an established risk factor for gastric cancer. This inhibitory effect was observed irrespective of the antibiotic resistance status of the bacterium. Another study found that H. pylori colonization in mice and humans was alleviated after a two-month feeding of broccoli sprouts.128 In addition to direct antibacterial effects on H. pylori, cytoprotective responses in human cells may play a role, as the protective effects of ITCs were not observed in Nrf2 knockout mice. The transcription factor Nrf2 is involved in the induction of a battery of antioxidant and phase 2 biotransformation enzymes.37–41 These enzymes protect the mucosal cells from oxidative stress and subsequent DNA damage.129 ITCs have also been shown to have bactericidal effects on food borne pathogens, including Escherichia coli,130Pseudomonas aeruginosa, Listeria monocytogenes and Staphylococcus aureus,131 supporting a broader anti-bacterial effect for these compounds.Given their anti-bacterial effects, ITCs may also modulate the human gut microbial community. Glucosinolates reach the large intestine and gut bacteria play an important role in metabolizing these dietary constituents to ITCs and other compounds in vivo when cruciferous vegetables are ingested.6,132 Aires et al.133 examined the effects of four ITCs on 17 human gut-associated bacteria. All ITCs had some anti-bacterial effects and SFN and BITC were the strongest inhibitors. These effects were dose-dependent with the strongest effects observed at high concentrations (3 μM). In humans, Shapiro et al.132 reported that urinary ITC excretion with cruciferous vegetable consumption decreased significantly when participants were pretreated with antibiotics and bowel cleansing. Further details on the anti-bacterial effects of ITCs can be found in other publications.134,135
Interindividual variation in biologic response in humans
By way of necessity, much of the mechanistic work on ITCs has been conducted in vitro, in cell systems, or in well-controlled animal models using isolated compounds. In humans consuming cruciferous vegetables, biologic response may vary due to differences in types and amounts of crucifers consumed, as well as genetic and other factors that influence exposure, metabolism and disposition of the ITCs, and interaction of the ITCs with target genes. Exposure to ITCs in vivo may be influenced by the environment in the digestive tract (e.g., hydrolysis by gut microbiota, microbiota composition, pH, nutrient interactions, etc.), how well food is chewed, and genetic variation in enzymes involved in metabolism of ITCs.47 These factors may translate into interindividual differences in the protective effects of cruciferous vegetables.ITCs are metabolized by GST through the addition of a glutathione moiety, producing dithiocarbamates; then they are further metabolized to N-acetylcysteine conjugates and broken down via the mercapturic acid pathway.136 Null genotypes of GSTM1 and GSTT1 result in a complete lack of their respective enzymes and considerable variability in the modifying effect of GST genotypes on cancer risk is observed.137–144 Based on pharmacokinetic studies, this variability does not appear to be the result of slower ITC metabolism, as ITCs appear to be excreted at a greater rate among GSTM1-null individuals.44,145 This observation may reflect ITC regulation of other signaling pathways that affect GST activity or differences in the type or amounts of ITCs consumed. It may also be that variants in N-acetyltransferase enzymes which produce N-acetylcysteine conjugates, further down the ITC metabolic pathway after conjugation with glutathione, are responsible, in part, for these observed differences.146
As discussed in the context of the anti-bacterial effects of ITCs, metabolism by gut bacteria may be another source of variation in ITC exposure. Typically, most crucifers eaten by humans are consumed cooked, and plant myrosinase is largely destroyed. In in vitro incubations of fecal or pure bacterial samples with select glucosinolates, several gut bacterial species have been shown to degrade glucosinolates.147–150 Feeding studies have also shown significant interindividual differences in urinary ITC recovery (1%–50% of original glucosinolate ingested) after participants consumed the same amount of cooked cruciferous vegetables.132,151–155 The variation was smaller when raw vegetables or ITCs were consumed directly, indicating individual differences in the ability to metabolize glucosinolates.132,151–155 A recent study by our group showed that fecal bacteria obtained from persons who had higher urinary ITC excretion after broccoli feeding degraded more glucoraphanin in vitro after a two day incubation compared to the bacteria from the low excreters. However, no overall fecal microbiota composition differences were detected between the two groups, possibly due to the complexity of micriobiota structure, and functional redundancy of glucosinolate metabolism within the community.156
Other factors in the digestive tract may also contribute to the variation of ITC exposure in vivo. For example, a small percentage of glucosinolates may undergo acidic hydrolysis in stomach.157 In addition, the presence of iron and acidic environment in the intestine were found to lead to nitrile formation instead of ITCs from glucosinolates.12,158
Summary and future directions
Accumulated evidence from studies conducted in cell culture, animal models, and epidemiologic cohorts over the past several decades have demonstrated an important role for ITCs in dietary prevention of several cancers. The biological effects of ITCs are diverse, involving multiple signaling pathways as well as cross-talk between pathways, and characterization of these continues to evolve. Given the variation in chemical structure of ITCs produced from different glucosinolates, this diversity is not unexpected. However, even with the wealth of information we have on the chemoprotective effects of ITCs to date, gaps in our understanding of their function remain.While there is value in elucidating the effects of ITCs on isolated pathways looking at a small number of targets, there is a need to understand systemic response of cells to ITCs from a whole-body perspective. Use of a systems biology approach, employing metagenomic, transcriptomic, proteomic and metabolomic technology may offer more comprehensive information on the actions of ITCs. For example, Traka et al.159 examined gene expression profiles in prostate tissue before and after men consumed 400 g broccoli per week for 12 months. Using pathway analyses, they found diet-induced changes in insulin signaling, transforming growth factor-β1 and epidermal growth factor signaling pathways. Interestingly, these changes were greater among carriers of a GSTM1+ genotype.159 We recently utilized high throughput proteomics methods to determine how human serum peptides changed in response to cruciferous vegetables in a controlled feeding trial. We reported significant changes in circulating levels of several peptides, including GSTM1 genotype-dependent decreases in transthyretin (TTR), a carrier protein for retinol and the thyroid hormone thyroxine, and zinc α-glycoprotein, an adipokine involved in lipid metabolism.160
These previously unrecognized responses to cruciferous vegetables point to the complexity of the mechanisms involved in crucifer-mediated effects, and modulation by GST genotype. Further, the effects of dose, duration of exposure, specific ITCs, and other host genetic factors should be considered. Because precursor glucosinolates, rather than ITCs, are present in cruciferous vegetables, investigation of glucosinolate metabolism by gut microbiota may also be an important link between cruciferous vegetable consumption with ITC exposure. Finally, in addition to systems biology approaches, future investigations should examine the effects of ITCs in vivo, in humans, through study of intermediate biomarkers of cancer-related processes.
References
- A. S. Keck and J. W. Finley, Integr. Cancer Ther., 2004, 3, 5–12 CrossRef CAS.
- International Agency for Research on Cancer, Cruciferous Vegetables, Isothiocyanates and Indoles, International Agency for Research on CancerLyon France, 2004 Search PubMed.
- G. Murillo and R. G. Mehta, Nutr. Cancer, 2001, 41, 17–28 CAS.
- J. H. Cohen, A. R. Kristal and J. L. Stanford, J. Natl. Cancer Inst., 2000, 92, 61–68 CrossRef CAS.
- K. A. Steinmetz and J. D. Potter, J. Am. Diet. Assoc., 1996, 96, 1027–1039 CrossRef CAS.
- T. A. Shapiro, J. W. Fahey, A. T. Dinkova-Kostova, W. D. Holtzclaw, K. K. Stephenson, K. L. Wade, L. Ye and P. Talalay, Nutr. Cancer, 2006, 55, 53–62 CrossRef CAS.
- M. M. Kushad, A. F. Brown, A. C. Kurilich, J. A. Juvik, B. P. Klein, M. A. Wallig and E. H. Jeffery, J. Agric. Food Chem., 1999, 47, 1541–1548 CrossRef CAS.
- R. F. Mithen, M. Dekker, R. Verkerk, S. Rabot and I. T. Johnson, J. Sci. Food Agric., 2000, 80, 967–984 CrossRef CAS.
- S. S. Hecht, S. G. Carmella, P. M. Kenney, S. H. Low, K. Arakawa and M. C. Yu, Cancer Epidemiol. Biomarkers Prev., 2004, 13, 997–1004 CAS.
- M. Vermeulen, R. Van den Berg, A. P. Freidig, P. J. Van Bladeren and W. H. J. Vaes, J. Agric. Food Chem., 2006, 54, 5350–5358 CrossRef CAS.
- J. W. Fahey, A. T. Zalcmann and P. Talalay, Phytochemistry, 2001, 56, 5–51 CrossRef CAS.
- D. J. Williams, C. Critchley, S. Pun, M. Chaliha and T. J. O'Hare, Phytochemistry, 2009, 70, 1401–1409 CrossRef CAS.
- Y. Zhang and P. Talalay, Cancer Res., 1998, 58, 4632–4639 CAS.
- L. Ye and Y. Zhang, Carcinogenesis, 2001, 22, 1987–1992 CrossRef CAS.
- M. Vermeulen, A. M. Boerboom, B. M. Blankvoort, J. M. Aarts, I. M. Rietjens, P. J. van Bladeren and W. H. Vaes, Toxicol. in Vitro, 2009, 23, 617–621 CrossRef CAS.
- J. Jakubikova, Y. Bao and J. Sedlak, Anticancer Res., 2005, 25, 3375–3386 CAS.
- R. Munday, Y. Zhang, C. M. Munday, M. V. Bapardekar and J. D. Paonessa, Pharm. Res., 2008, 25, 2164–2170 CrossRef CAS.
- A. Prawan, C. L. Saw, T. O. Khor, Y. S. Keum, S. Yu, L. Hu and A. N. Kong, Chem.-Biol. Interact., 2009, 179, 202–211 CrossRef CAS.
- M. Lopez and L. Mazzanti, J. Pathol. Bacteriol., 1955, 69, 243–250 CrossRef CAS.
- L. M. Mc and K. R. Rees, J. Pathol. Bacteriol., 1958, 76, 175–188 CrossRef.
- L. W. Wattenberg, J. Natl. Cancer Inst., 1977, 58, 395–398 CAS.
- S. S. Hecht, Drug Metab. Rev., 2000, 32, 395–411 CrossRef CAS.
- Y. Zhang and P. Talalay, Cancer Res., 1994, 54, 1976s–1981s CAS.
- J. D. Hayes, M. O. Kelleher and I. M. Eggleston, Eur. J. Nutr., 2008, 47(S2), 73–88 CrossRef CAS.
- D. M. Minich and J. S. Bland, Nutr. Rev., 2008, 65, 259–267 CrossRef.
- J. V. Higdon, B. Delage, D. E. Williams and R. H. Dashwood, Pharmacol. Res., 2007, 55, 224–236 CrossRef CAS.
- C. W. Nho and E. Jeffery, Toxicol. Appl. Pharmacol., 2004, 198, 40–48 CrossRef CAS.
- L. W. Wattenberg, Cancer Res., 1981, 41, 2991–2994 CAS.
- L. W. Wattenberg, Proc. Nutr. Soc., 2007, 49, 173–183 CrossRef.
- P. Talalay, J. W. Fahey, W. D. Holtzclaw, T. Prestera and Y. Zhang, Toxicol. Lett., 1995, 82–83, 173–179 CrossRef CAS.
- D. L. Eaton and T. K. Bammler, Toxicol. Sci., 1999, 49, 156–164 CrossRef CAS.
- A. T. Dinkova-Kostova, W. D. Holtzclaw, R. N. Cole, K. Itoh, N. Wakabayashi, Y. Katoh, M. Yamamoto and P. Talalay, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11908–11913 CrossRef CAS.
- J. D. Hayes and M. McMahon, Trends Biochem. Sci., 2009, 34, 176–188 CrossRef CAS.
- P. Talalay, BioFactors, 2000, 12, 5–11 CrossRef CAS.
- G. P. Basten, Y. Bao and G. Williamson, Carcinogenesis, 2002, 23, 1399–1404 CrossRef CAS.
- X. Gao and P. Talalay, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10446–10451 CrossRef CAS.
- S. L. Slocum and T. W. Kensler, Arch. Toxicol., 2011, 85, 273–284 CrossRef CAS.
- Y. J. Surh, J. K. Kundu and H. K. Na, Planta Med., 2008, 74, 1526–1539 CrossRef CAS.
- L. Valgimigli and R. Iori, Environ. Mol. Mutagen., 2009, 50, 222–237 CrossRef CAS.
- Y. Nakamura and N. Miyoshi, Biosci., Biotechnol., Biochem., 2010, 74, 242–255 CrossRef CAS.
- L. Baird and A. T. Dinkova-Kostova, Arch. Toxicol., 2011, 85, 241–272 CrossRef CAS.
- S. L. Navarro, S. Peterson, C. Chen, K. W. Makar, Y. Schwarz, I. B. King, S. S. Li, L. Li, M. Kestin and J. W. Lampe, Cancer Prev. Res., 2009, 2, 345–352 CrossRef CAS.
- S. L. Navarro, J. Chang, S. Peterson, C. Chen, I. B. King, Y. Schwarz, S. S. Li, J. D. Potter and J. W. Lampe, Cancer Epidemiol., Biomarkers Prev., 2009, 18, 2974–2978 CrossRef CAS.
- A. V. Gasper, A. Al-Janobi, J. A. Smith, J. R. Bacon, P. Fortun, C. Atherton, M. A. Taylor, C. J. Hawkey, D. A. Barrett and R. F. Mithen, Am. J. Clin. Nutr., 2005, 82, 1283–1291 CAS.
- C. C. Conaway, Y. M. Yang and F. L. Chung, Curr. Drug Metab., 2002, 3, 233–255 CrossRef CAS.
- D. T. Verhoeven, H. Verhagen, R. A. Goldbohm, P. A. van den Brandt and G. van Poppel, Chem.-Biol. Interact., 1997, 103, 79–129 CrossRef CAS.
- J. W. Lampe and S. Peterson, J. Nutr., 2002, 132, 2991–2994 CAS.
- P. Talalay and J. W. Fahey, J. Nutr., 2001, 131, 3027S–3033S CAS.
- M. C. Myzak and R. H. Dashwood, Cancer Lett., 2006, 233, 208–218 CrossRef CAS.
- J. D. Clarke, R. H. Dashwood and E. Ho, Cancer Lett., 2008, 269, 291–304 CrossRef CAS.
- N. Juge, R. F. Mithen and M. Traka, Cell. Mol. Life Sci., 2007, 64, 1105–1127 CrossRef CAS.
- Y. S. Keum, W. S. Jeong and A. N. T. Kong, Mutat. Res., 2004, 555, 191–202 CrossRef CAS.
- H. L. Pahl, Oncogene, 1999, 18, 6853–6866 CrossRef CAS.
- E. Rath and D. Haller, Eur. J. Nutr., 2011, 50, 219–233 CrossRef CAS.
- T. Sathyapalan and S. L. Atkin, Minerva Endocrinol., 2011, 36, 147–156 CAS.
- B. H. Maskrey, I. L. Megson, P. D. Whitfield and A. G. Rossi, Arterioscler., Thromb., Vasc. Biol., 2011, 31, 1001–1006 CrossRef CAS.
- S. I. Grivennikov and M. Karin, Ann. Rheum. Dis., 2011, 70(Suppl 1), i104–108 CrossRef CAS.
- S. Nair, W. Li and A. N. Kong, Acta Pharmacol. Sin., 2007, 28, 459–472 CrossRef CAS.
- T. D. Gilmore and M. Herscovitch, Oncogene, 2006, 25, 6887–6899 CrossRef CAS.
- W. S. Jeong, I. W. Kim, R. Hu and A. N. Kong, Pharm. Res., 2004, 21, 661–670 CrossRef CAS.
- H. S. Youn, Y. S. Kim, Z. Y. Park, S. Y. Kim, N. Y. Choi, S. M. Joung, J. A. Seo, K. M. Lim, M. K. Kwak, D. H. Hwang and J. Y. Lee, J. Immunol., 2009, 184, 411–419 CrossRef.
- L. Wu, M. H. Noyan Ashraf, M. Facci, R. Wang, P. G. Paterson, A. Ferrie and B. H. Juurlink, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7094–7099 CrossRef CAS.
- H. Ohshima and H. Bartsch, Mutat. Res., 1994, 305, 253–264 CrossRef CAS.
- Z. Culig, Biochim. Biophys. Acta, Mol. Cell Res., 2011, 1813, 308–314 CrossRef CAS.
- A. Mantovani, P. Allavena, A. Sica and F. Balkwill, Nature, 2008, 454, 436–444 CrossRef CAS.
- Y. S. Keum, W. S. Jeong and A. N. Kong, Drug News Perspect., 2005, 18, 445–451 CAS.
- K. L. Cheung and A. N. Kong, AAPS J., 2009, 12, 87–97 CrossRef.
- T. D. Gilmore, Oncogene, 2006, 25, 6680–6684 CrossRef CAS.
- M. Karin, Nature, 2006, 441, 431–436 CrossRef CAS.
- E. Heiss, C. Herhaus, K. Klimo, H. Bartsch and C. Gerhauser, J. Biol. Chem., 2001, 276, 32008–32015 CrossRef CAS.
- W. Li, T. O. Khor, C. Xu, G. Shen, W. S. Jeong, S. Yu and A. N. Kong, Biochem. Pharmacol., 2008, 76, 1485–1489 CrossRef CAS.
- Y. J. Surh and H. K. Na, Genes Nutr., 2007, 2, 313–317 CrossRef.
- M. Yu, H. Li, Q. Liu, F. Liu, L. Tang, C. Li, Y. Yuan, Y. Zhan, W. Xu, W. Li, H. Chen, C. Ge, J. Wang and X. Yang, Cell. Signalling, 2011, 23, 883–892 CrossRef CAS.
- S. Nair, S. T. Doh, J. Y. Chan, A. N. Kong and L. Cai, Br. J. Cancer, 2008, 99, 2070–2082 CrossRef CAS.
- T. O. Khor, M. T. Huang, K. H. Kwon, J. Y. Chan, B. S. Reddy and A. N. Kong, Cancer Res., 2006, 66, 11580–11584 CrossRef CAS.
- Q. Ma, L. Battelli and A. F. Hubbs, Am. J. Pathol., 2006, 168, 1960–1974 CrossRef CAS.
- K. Yoh, K. Itoh, A. Enomoto, A. Hirayama, N. Yamaguchi, M. Kobayashi, N. Morito, A. Koyama, M. Yamamoto and S. Takahashi, Kidney Int., 2001, 60, 1343–1353 CrossRef CAS.
- G. H. Liu, J. Qu and X. Shen, Biochim. Biophys. Acta, Mol. Cell Res., 2008, 1783, 713–727 CrossRef CAS.
- H. Nian, B. Delage, E. Ho and R. H. Dashwood, Environ. Mol. Mutagen., 2009, 50, 213–221 CrossRef CAS.
- R. H. Dashwood and E. Ho, Semin. Cancer Biol., 2007, 17, 363–369 CrossRef CAS.
- L. G. Wang and J. W. Chiao, Int. J. Oncol., 2010, 37, 533–539 CAS.
- R. H. Dashwood and E. Ho, Nutr. Rev., 2008, 66(Suppl. 1), S36–38 CrossRef.
- M. F. Fraga and M. Esteller, Cell Cycle, 2005, 4, 1377–1381 CrossRef CAS.
- M. C. Myzak, K. Hardin, M. Yan, P. Tong, R. Dashwood and E. Ho, FASEB J., 2006, 20, A150–A150 Search PubMed.
- M. A. Lea, M. Rasheed, V. M. Randolph, F. Khan, A. Shareef and C. desBordes, Nutr. Cancer, 2002, 43, 90–102 CrossRef CAS.
- M. C. Myzak and R. H. Dashwood, Curr. Drug Targets, 2006, 7, 443–452 CrossRef CAS.
- S. Batra, R. P. Sahu, P. K. Kandala and S. K. Srivastava, Mol. Cancer Ther., 2010, 9, 1596–1608 CrossRef CAS.
- Q. Lu, X. Lin, J. Feng, X. Zhao, R. Gallagher, M. Y. Lee, J. W. Chiao and D. Liu, J. Hematol. Oncol., 2008, 1, 6 CrossRef.
- M. C. Myzak, K. Hardin, R. Wang, R. H. Dashwood and E. Ho, Carcinogenesis, 2005, 27, 811–819 CrossRef.
- C. Fimognari and P. Hrelia, Mutat. Res., Rev. Mutat. Res., 2007, 635, 90–104 CrossRef CAS.
- S. M. Meeran, S. N. Patel and T. O. Tollefsbol, PLoS One, 2010, 5, e11457 Search PubMed.
- L. G. Wang, A. Beklemisheva, X. M. Liu, A. C. Ferrari, J. Feng and J. W. Chiao, Mol. Carcinog., 2007, 46, 24–31 CrossRef CAS.
- D. P. Bartel, Cell, 2009, 136, 215–233 CrossRef CAS.
- A. Izzotti, P. Larghero, C. Cartiglia, M. Longobardi, U. Pfeffer, V. E. Steele and S. De Flora, Carcinogenesis, 2010, 31, 894–901 CrossRef CAS.
- A. Izzotti, G. A. Calin, V. E. Steele, C. Cartiglia, M. Longobardi, C. M. Croce and S. De Flora, Cancer Prev. Res., 2010, 3, 62–72 CrossRef CAS.
- A. V. Singh, D. Xiao, K. L. Lew, R. Dhir and S. V. Singh, Carcinogenesis, 2003, 25, 83–90 CrossRef.
- D. Xiao, C. S. Johnson, D. L. Trump and S. V. Singh, Mol. Cancer Ther., 2004, 3, 567–575 CAS.
- N. G. Chen, K. T. Chen, C. C. Lu, Y. H. Lan, C. H. Lai, Y. T. Chung, J. S. Yang and Y. C. Lin, Oncol. Rep., 2010, 24, 449–455 CAS.
- L. Mi, N. Gan and F. L. Chung, Carcinogenesis, 2010, 32, 216–223 CrossRef.
- R. Hu, T. O. Khor, G. Shen, W. S. Jeong, V. Hebbar, C. Chen, C. Xu, B. Reddy, K. Chada and A. N. Kong, Carcinogenesis, 2006, 27, 2038–2046 CrossRef CAS.
- S. Y. Park, G. Y. Kim, S. J. Bae, Y. H. Yoo and Y. H. Choi, Oncol. Rep., 2007, 18, 181–187 CAS.
- S. Choi and S. V. Singh, Cancer Res., 2005, 65, 2035–2043 CrossRef CAS.
- D. Xiao, A. A. Powolny, M. B. Moura, E. E. Kelley, A. Bommareddy, S. H. Kim, E. R. Hahm, D. Normolle, B. Van Houten and S. V. Singh, J. Biol. Chem., 2010, 285, 26558–26569 CrossRef CAS.
- J. D. Clarke, A. Hsu, Z. Yu, R. H. Dashwood and E. Ho, Mol. Nutr. Food Res., 2011 Search PubMed.
- L. Gamet-Payrastre, Curr. Cancer Drug Targets, 2006, 6, 135–145 CrossRef CAS.
- Y. Nakamura, Forum Nutr., 2009, 61, 170–181 CrossRef CAS.
- Y. Zhang, S. Yao and J. Li, Proc. Nutr. Soc., 2007, 65, 68–75 CrossRef.
- X. Wu, Q. H. Zhou and K. Xu, Acta Pharmacol. Sin., 2009, 30, 501–512 CrossRef CAS.
- C. A. Heinlein and C. Chang, Endocr. Rev., 2004, 25, 276–308 CrossRef CAS.
- B. J. Deroo and K. S. Korach, J. Clin. Invest., 2006, 116, 561–570 CAS.
- Y. S. Kim and J. A. Milner, J. Nutr. Biochem., 2005, 16, 65–73 CrossRef CAS.
- A. A. Beklemisheva, J. Feng, Y. A. Yeh, L. G. Wang and J. W. Chiao, Prostate, 2007, 67, 863–870 CrossRef CAS.
- L. Kang, L. Ding and Z. Y. Wang, Oncol Rep, 2009, 21, 185–192 CAS.
- M. C. Ramirez and K. Singletary, J. Nutr. Biochem., 2009, 20, 195–201 CrossRef CAS.
- U. Telang, D. A. Brazeau and M. E. Morris, Exp. Biol. Med., 2009, 234, 287–295 CrossRef CAS.
- K. Paech, P. Webb, G. G. Kuiper, S. Nilsson, J. Gustafsson, P. J. Kushner and T. S. Scanlan, Science, 1997, 277, 1508–1510 CrossRef CAS.
- B. E. Cavell, S. S. Syed Alwi, A. Donlevy and G. Packham, Biochem. Pharmacol., 2011, 81, 327–336 CrossRef CAS.
- S. J. Jackson and K. W. Singletary, J. Nutr., 2004, 134, 2229–2236 CAS.
- T. K. Smith, E. K. Lund, M. L. Parker, R. G. Clarke and I. T. Johnson, Carcinogenesis, 2004, 25, 1409–1415 CrossRef CAS.
- L. Mi, N. Gan, A. Cheema, S. Dakshanamurthy, X. Wang, D. C. Yang and F. L. Chung, J. Biol. Chem., 2009, 284, 17039–17051 CrossRef CAS.
- P. Yin, T. Kawamura, M. He, D. K. Vanaja and C. Y. Young, Cell Biol. Int., 2009, 33, 57–64 CrossRef CAS.
- E. S. Hwang and H. J. Lee, Exp. Biol. Med. (Maywood), 2006, 231, 421–430 CAS.
- P. Thejass and G. Kuttan, Immunopharmacol. Immunotoxicol., 2006, 28, 443–457 CrossRef CAS.
- L. Tang, G. R. Zirpoli, K. Guru, K. B. Moysich, Y. Zhang, C. B. Ambrosone and S. E. McCann, Cancer Epidemiol., Biomarkers Prev., 2010, 19, 1806–1811 CrossRef CAS.
- T. A. Dolecek, B. J. McCarthy, C. E. Joslin, C. E. Peterson, S. Kim, S. A. Freels and F. G. Davis, J. Am. Diet. Assoc., 2010, 110, 369–382 CrossRef.
- R. Chan, K. Lok and J. Woo, Mol. Nutr. Food Res., 2009, 53, 201–216 CAS.
- J. W. Fahey, X. Haristoy, P. M. Dolan, T. W. Kensler, I. Scholtus, K. K. Stephenson, P. Talalay and A. Lozniewski, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7610–7615 CrossRef CAS.
- A. Yanaka, J. W. Fahey, A. Fukumoto, M. Nakayama, S. Inoue, S. Zhang, M. Tauchi, H. Suzuki, I. Hyodo and M. Yamamoto, Cancer Prev. Res., 2009, 2, 353–360 CrossRef CAS.
- A. Yanaka, Curr. Pharm. Des., 2011 Search PubMed.
- F. B. Luciano and R. A. Holley, Int. J. Food Microbiol., 2009, 131, 240–245 CrossRef CAS.
- M. J. Saavedra, A. Borges, C. Dias, A. Aires, R. N. Bennett, E. S. Rosa and M. Simoes, Med. Chem., 2010, 6, 174–183 CAS.
- T. A. Shapiro, J. W. Fahey, K. L. Wade, K. K. Stephenson and P. Talalay, Cancer Epidemiol. Biomarkers Prev., 1998, 7, 1091–1100 CAS.
- A. Aires, V. R. Mota, M. J. Saavedra, E. A. Rosa and R. N. Bennett, J. Appl. Microbiol., 2009, 106, 2086–2095 CrossRef CAS.
- J. K. Moon, J. R. Kim, Y. J. Ahn and T. Shibamoto, J. Agric. Food Chem., 2010, 58, 6672–6677 CrossRef CAS.
- X. Haristoy, J. W. Fahey, I. Scholtus and A. Lozniewski, Planta Med., 2005, 71, 326–330 CrossRef CAS.
- R. H. Kolm, H. Danielson, Y. Zhang, P. Talalay and B. Mannervik, Biochem. J., 1995, 311, 453–459 CAS.
- S. J. London, J. M. Yuan, F. L. Chung, Y. T. Gao, G. A. Coetzee, R. K. Ross and M. C. Yu, Lancet, 2000, 356, 724–729 CrossRef CAS.
- B. Zhao, A. Seow, E. J. D. Lee, W.-T. Poh, M. Teh, P. Eng, Y.-T. Wang, W.-C. Tan, M. C. Yu and H.-P. Lee, Cancer Epidemiol. Biomarkers Prev., 2001, 10, 1063–1067 CAS.
- A. Seow, J. M. Yuan, C. L. Sun, D. Van Den Berg, H. P. Lee and M. C. Yu, Carcinogenesis, 2002, 23, 2055–2061 CrossRef CAS.
- J. H. Fowke, X. O. Shu, Q. Dai, A. Shintani, C. C. Conaway, F. L. Chung, Q. Cai, Y. T. Gao and W. Zheng, Cancer Epidemiol. Biomarkers Prev., 2003, 12, 1536–1539 CAS.
- M. R. Spitz, C. M. Duphorne, M. A. Detry, P. C. Pillow, C. I. Amos, L. Lei, M. de Andrade, X. Gu, W. K. Hong and X. Wu, Cancer Epidemiol. Biomarkers Prev., 2000, 9, 1017–1020 CAS.
- M. A. Joseph, K. B. Moysich, J. L. Freudenheim, P. G. Shields, E. D. Bowman, Y. Zhang, J. R. Marshall and C. B. Ambrosone, Nutr. Cancer, 2004, 50, 206–213 CrossRef CAS.
- L. I. Wang, E. L. Giovannucci, D. Hunter, D. Neuberg, L. Su and D. C. Christiani, Cancer, Causes Control, 2004, 15, 977–985 CrossRef.
- S. E. Steck, M. M. Gaudet, J. A. Britton, S. L. Teitelbaum, M. B. Terry, A. I. Neugut, R. M. Santella and M. D. Gammon, Carcinogenesis, 2007, 28, 1954–1959 CrossRef CAS.
- S. E. Steck, M. D. Gammon, J. R. Hebert, D. E. Wall and S. H. Zeisel, J. Nutr., 2007, 137, 904–909 CAS.
- R. Munday, P. Mhawech-Fauceglia, C. M. Munday, J. D. Paonessa, L. Tang, J. S. Munday, C. Lister, P. Wilson, J. W. Fahey, W. Davis and Y. Zhang, Cancer Res., 2008, 68, 1593–1600 CrossRef CAS.
- A. D. Brabban and C. Edwards, FEMS Microbiol. Lett., 1994, 119, 83–88 CrossRef CAS.
- S. Rabot, C. Guerin, L. Nugon-Boudon and O. Szylit, Proc 9th Int. Rapeseed Congress, 1995, 212–214 Search PubMed.
- L. Elfoul, S. Rabot, N. Khelifa, A. Quinsac, A. Duguay and A. Rimbault, FEMS Microbiol. Lett., 2001, 197, 99–103 CrossRef CAS.
- D. L. Cheng, K. Hashimoto and Y. Uda, Food Chem. Toxicol., 2004, 42, 351–357 CrossRef CAS.
- T. A. Shapiro, J. W. Fahey, K. L. Wade, K. K. Stephenson and P. Talalay, Cancer Epidemiol. Biomarkers Prev., 2001, 10, 501–508 CAS.
- S. M. Getahun and F. L. Chung, Cancer Epidemiol. Biomarkers Prev., 1999, 8, 447–451 CAS.
- C. C. Conaway, S. M. Getahun, L. L. Liebes, D. J. Pusateri, D. K. W. Topham, M. Botero-Omary and F. L. Chung, Nutr. Cancer, 2000, 38, 168–178 CrossRef CAS.
- G. Rouzaud, S. A. Young and A. J. Duncan, Cancer Epidemiol., Biomarkers Prev., 2004, 13, 125–131 CAS.
- F. Li, M. A. Hullar, S. A. Beresford and J. W. Lampe, Br. J. Nutr., 2011, 106, 408–416 CrossRef CAS.
- F. Li, M. A. Hullar, Y. Schwarz and J. W. Lampe, J. Nutr., 2009, 139, 1685–1691 CrossRef CAS.
- H. G. Tiedink, L. H. de Haan, W. M. Jongen and J. H. Koeman, Cell Biol. Toxicol., 1991, 7, 371–386 CrossRef CAS.
- L. Ludikhuyze, L. Rodrigo and M. Hendrickx, J. Food Prot., 2000, 63, 400–403 CAS.
- M. Traka, A. V. Gasper, A. Melchini, J. R. Bacon, P. W. Needs, V. Frost, A. Chantry, A. M. Jones, C. A. Ortori, D. A. Barrett, R. Y. Ball, R. D. Mills and R. F. Mithen, PLoS One, 2008, 3, e2568 Search PubMed.
- H. A. Brauer, T. E. Libby, B. L. Mitchell, L. Li, C. Chen, T. W. Randolph, Y. Y. Yasui, J. W. Lampe and P. D. Lampe, Nutr. J., 2011, 10, 11 CrossRef.
Footnote |
| † Sponsorship: This work was supported by grants R01CA142695, R56CA70913, and R25CA94880 from the National Institutes of Health, National Cancer Institute. |
| This journal is © The Royal Society of Chemistry 2011 |