Naturally occurring inhibitors against the formation of advanced glycation end-products
Xiaofang
Peng
a,
Jinyu
Ma
a,
Feng
Chen
ab and
Mingfu
Wang
*a
aSchool of Biological Sciences, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China. E-mail: mfwang@hkusua.hku.hk; Fax: +22990340; Tel: +22990338
bInstitute for Food & Bioresource Engineering, College of Engineering, Peking University, Beijing, P. R. China
First published on 15th June 2011
Abstract
Advanced glycation end-products (AGEs) are the final products of the non-enzymatic reaction between reducing sugars and amino groups in proteins, lipids and nucleic acids. Recently, the accumulation of AGEs in vivo has been implicated as a major pathogenic process in diabetic complications, atherosclerosis, Alzheimer's disease and normal aging. The early recognition of AGEs can ascend to the late 1960s when a non-enzymatic glycation process was found in human body which is similar to the Maillard reaction. To some extent, AGEs can be regarded as products of the Maillard reaction. This review firstly introduces the Maillard reaction, the formation process of AGEs and harmful effects of AGEs to human health. As AGEs can cause undesirable diseases or disorders, it is necessary to investigate AGE inhibitors to offer a potential therapeutic approach for the prevention of diabetic or other pathogenic complications induced by AGEs. Typical effective AGE inhibitors with different inhibition mechanisms are also reviewed in this paper. Both synthetic compounds and natural products have been evaluated as inhibitors against the formation of AGEs. However, considering toxic or side effects of synthetic molecules present in clinical trials, natural products are more promising to be developed as potent AGE inhibitors.
1 Introduction
Advanced glycation end-products (AGEs) are the final products of a series of chemical reactions initiated by the attachment of reducing sugars to free amino groups in proteins, lipids and nucleic acids.1 The early recognition of AGEs can ascend to the late 1960s when a non-enzymatic glycation process similar to the Maillard reaction2,3 was found in human body by the observation of increased formation of glycosylated haemoglobins (AGE precursors) in diabetic patients.4,5 Later on, more than a dozen of AGEs have been determined in different tissues, such as retinal blood vessel walls, arterial collagen and renal tissues,6 which were formed at a slow but constant rate in the normal body, beginning from early embryonic development, and accumulating with time. Besides, AGEs were also widely detected in different food matrixes including bakery products and milk, formed through heating sugars together with proteins and other food ingredients.7AGEs are commonly viewed as a group of complex and heterogeneous compounds. Recently, their accumulation in vivo has been implicated as a major pathogenic process in diabetic complications, including neuropathy, nephropathy, retinopathy and cataract6,8 and other health disorders, such as atherosclerosis,9 Alzheimer's disease10,11 and normal aging.11,12 Meanwhile, some highly reactive carbonyl species (RCS) formed during the formation of AGEs are also believed to play an important role in diabetic complications. Therefore, the discovery and investigation of AGE inhibitors would offer a potential preventive and therapeutic approach for lowering the risks of diabetic or other pathogenic complications caused by AGE formation.
So far, both synthetic compounds and natural products have been evaluated as inhibitors against the formation of AGEs. Although some synthetic molecules have demonstrated strong power to inhibit the formation of AGEs or break AGE-induced crosslinks, they might also cause severe side effects as well.13–15 When compared to synthetic compounds, natural occurring products have been proven relatively safer for human consumption. In this regard, some plant extracts and their relevant phenolic ingredients have been evaluated for their effects on the formation of AGEs in recent years,16–19 and showed potent anti-glycative effects which are mainly due to their strong antioxidant activities. Considering aspects of safety and efficacy, natural products with strong inhibitory effects on AGE formation have great potential to be further investigated for the future application as functional foods or even as preventive drugs to fight against AGE-associated diseases and disorders.
2 Maillard reaction
In 1912, Louis Camille Maillard firstly discovered that sugars could react with amino acids under heating conditions and yield a yellow-brown color.20 The sugar-amino acid reaction later named after the chemist Maillard, is extraordinarily complicated. It was further elucidated that the Maillard reaction, initiated by the carbonyl group of a reducing sugar (such as glucose, fructose and lactose) reacting with the nucleophilic amino group of an amino acid, peptide or protein, could produce a large number of poorly characterized compounds that contribute to a series of odors and flavors. Meanwhile, different amino acids correspondingly create distinctive flavors. To some extent, the Maillard reaction is the foundation of the modern flavor industry. In addition, the Maillard reaction is also called non-enzymatic browning, being one of the main reasons for the occurrence of the non-enzymatic food browning. Thus, the Maillard reaction is of vital importance to food science.2,32.1 The process of the Maillard reaction
Generally speaking, the process of the Maillard reaction can be roughly divided into three main stages. The initial step of the non-enzymatic glycation is the covalent attachment of reducing sugars, such as glucose, fructose, or glucose-6-phosphate, to N-terminal amino acid residues or epsilon amino groups of proteins, lipids, and nucleic acids, which produces a Schiff base, a reversible and unstable N-substituted glycosylamine (Fig.1). In this phase, glucose shows the slowest glycation rate when compared to other reducing sugars.21,22 Subsequently, the Schiff base would experience a further isomerization called an Amadori rearrangement23 and change to more stable Amadori adducts, namely ketosamines (Fig. 2). The ketosamine products then undergo further dehydration either to form reductones and dehydro reductones, or to change to short chain hydrolytic fission products such as diacetyl, acetol or pyruvaldehyde. These carbonyl compounds can further engage in the Strecker degradation, giving rise to a series of additional aroma compounds (Fig. 3). At the same time, intermediates formed in the Maillard reaction including dehyro reductones, fission products and products of Strecker degradation could also take part in aldol condensation and aldehyde-amine condensation, leading to the formation of both brown nitrogen-containing pigments (melanoidins) and mutagenic heterocyclic nitrogen-containing compounds.3,24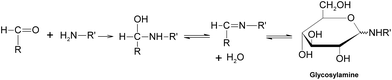 | ||
| Fig. 1 Formation of a Schiff base. | ||
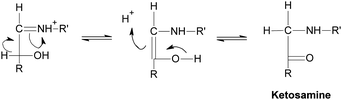 | ||
| Fig. 2 Amadori rearrangement. | ||
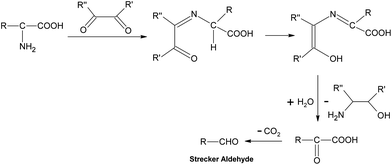 | ||
| Fig. 3 Strecker degradation. | ||
2.2 Effects of Maillard reaction
Color formation is an important characteristic of the Maillard reaction. The most studied colored products of the Maillard reaction are melanoidins which are brown, high molecular weight pigments, widely present in many foods like coffee, bread and beer. Melanoidins may have some beneficial antioxidant activities.25 However, up to now the knowledge about the chemical nature, functional and physiological properties of the colored moieties is still relatively limited. The color development caused by the Maillard reaction has both good and bad sides. For example, color development in meats, syrup and bread baking is desirable while the browning of dry milk or color changes in dehydrated products during storage is unfavorable. On the other hand, the Maillard reaction also gives rise to different volatile compounds, such as furans, pyrroles and pyridines, thus resulting in the enticing smell of coffee, breads and meats. Some researchers have summarized odor types from more than 400 Maillard models by using different amino acids as reactants. For example, aliphatic amino acids are essential for caramel aroma and sulfur-containing amino acids are required for meaty smell.26 However, when appreciating benefits brought by the Maillard reaction, we should also pay attention to its undesirable effects. From the nutritional aspects, the Maillard reaction may reduce the nutritional value of foods by losing some essential amino acids and carbohydrates which are potential reactants for the Maillard reaction. Meanwhile, the Maillard reaction can affect the activity of enzymes through glycation.27 Moreover, some of the Maillard reaction products have been found to be toxic. One of the well-known toxic Maillard products is acrylamide, which is a carcinogen formed in heated starch-riched foodstuffs, especially in French fries.28 Heterocyclic amines have also been reported to be carcinogenic and mutagenic products of the Maillard reaction, formed in fish and meats under heating conditions.29,30 In addition, non-enzymic glycosylation has been considered an important contributor to diabetic complications, which is due to the formation of advanced glycation end-products (AGEs). The following part will give a detailed introduction of AGEs.3 Advanced glycation end-products (AGEs)
Besides its wide application in the field of food science, the Maillard reaction also has physiological significance. In the late 1960s, a non-enzymatic glycation process similar to the Maillard reaction was recognized in the human body by the observation of increased formation of glycosylated haemoglobins in diabetic patients,4,5 which would lead to the formation of detrimental AGEs. AGEs are a group of complex and heterogeneous compounds which are known as brown and fluorescent cross-linking substances such as pentosidine, non-fluorescent cross-linking products such as glyoxal-lysine dimer (GOLD) and methylglyoxal-lysine dimer (MOLD), or non-fluorescent, non-crosslinking adducts such as carboxymethyllysine (CML) and pyrraline (a pyrrole aldehyde).31,32AGEs can be formed both in vivo through normal metabolism and in vitro by heating sugars together with fats or proteins.7 So far, more than a dozen of AGEs have been determined in different tissues.6 For examples, pyrraline was reported to be detected in brain tissue from patients with Alzheimer disease.33 Levels of both CML and pentosidine have been shown to increase in the skin collagen of diabetic patients.34,35 GOLD and MOLD were identified as major Maillard reaction cross-links in lens proteins and their concentrations were elevated with age.36 Meanwhile, AGEs were also reported to be determined in food matrixes. Dietary AGEs such as CML, pentosidine and pyralline, were commonly observed in bakery products and milk.37–39 As a common AGE marker, the highest levels of CML were found in foods with a high content of fats including roasted almonds, butters, olive oil and meats,40 but were also present in cola where pentosindine, GOLD and MOLD were determined as well.38 Whatever condition it is, the formation of AGEs is influenced by several factors, like sugar concentration, chemical structure, turnover rate of targeted protein and the degree of oxidative stress in the environment.41–44
3.1 Formation of general AGEs
Similar to the Maillard reaction process, AGE formation pathways start with the reaction between reducing sugars and amino groups. After the formation of Schiff base and the Amadori rearrangement, the Amadori products undergo subsequent dehydration and rearrangement to form highly reactive dicarbonyl compounds45 including 3-deoxyglucosone (3-DG), glyoxal (GO) and methylglyoxal (MGO) (Fig. 4). The reactions between these AGE precursors and amino, sulfhydryl and guanidine functional groups of intracellular and extracellular proteins36,46 would result in the formation of stable and irreversible AGEs (Fig. 5), which are composed of brown and fluorescent cross-linking compounds such as pentosidine, non-fluorescent cross-linking products such as methylglyoxal-lysine dimmers (MOLD, imidazolium salts), and also non-fluorescent, non-crosslinking adducts like carboxymethyllysine (CML) and pyrraline.31,32 Meanwhile, AGEs could also be formed by Amadori products directly through rearrangement under both oxidative and non-oxidative conditions.1 In addition, the reactive dicarbonyls can be generated from other pathways (Fig. 6). For example, Schiff base may engage in oxidative fragmentation to form such dicarbonyl intermediates (Namiki pathway).47 At the same time, metal-catalyzed auto-oxidation of glucose (Wolff pathway) and lipid peroxidation (Acetol pathway) are other sources of reactive AGE precursors.48,49 The increase in the concentration of reactive carbonyls both from oxidative and non-oxidative reactions, glycoxidation and lipoxidation products may result in carbonyl stress,50,51 which leads to increased chemical modifications of proteins and causes oxidative stress.52 AGE modification of proteins could further bring on a cascade of events including the activation of signal transduction pathways and subsequent activation of inflammatory responses, leading to tissue damage.53 Once formed, AGEs may continue to take part in a covalent cross-link with proteins which are generally stable and long lived, such as collagen and crystallins.45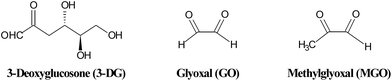 | ||
| Fig. 4 Structures of some reactive dicarbonyls formed during glycation. | ||
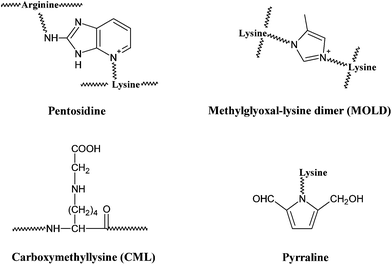 | ||
| Fig. 5 Structures of some typical AGEs. | ||
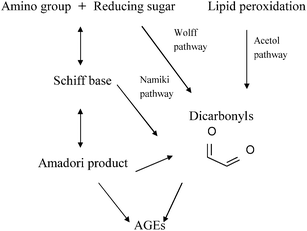 | ||
| Fig. 6 Glycation pathways of AGE formation. | ||
3.2 Formation of typical AGEs
So far, the knowledge of chemical and functional properties of most AGEs is relatively limited. The representatives of well-characterized and widely studied AGEs up to now may be pentosidine, CML and MGO derivatives, which have been detected in a range of human tissues and serve as biomarkers of AGEs, providing new insights into the biochemistry of AGE compounds in general.34,35 Pentosidine, as mentioned above, is a fluorescent cross-link formed in the greatest yield in reactions of pentoses with lysine and arginine residues.54–56 It can also be formed from other carbohydrate sources, such as glucose, fructose and ascorbate under aerobic conditions,56,57 with relatively lower yield (Fig. 7). Pentosidine was determined in different tissue proteins, such as lens proteins and skin collagens,58,59 accumulating with age and diabetes.60 It was reported that pentosidine was present in human tissues at only trace concentrations (approximately 25 μmol pentosidine/mol lens crystalline protein and 2 mmol pentosidine/mol collagen at age 70), and accounted for a small fraction of the total modification of lysine or arginine residues of proteins, suggesting that this level of pentosidine might not have a significant effect on the structure and function of relevant proteins.56 However, the kinetics of the formation and accumulation of pentosidine in tissues exactly reflect more extensive chemical modifications (such as loss of lysine or arginine residues, cross-linking of proteins) and possible damages to proteins. Hence, pentosidine should be considered as an important index for evaluating carbohydrate-dependant damages to proteins under glycation.61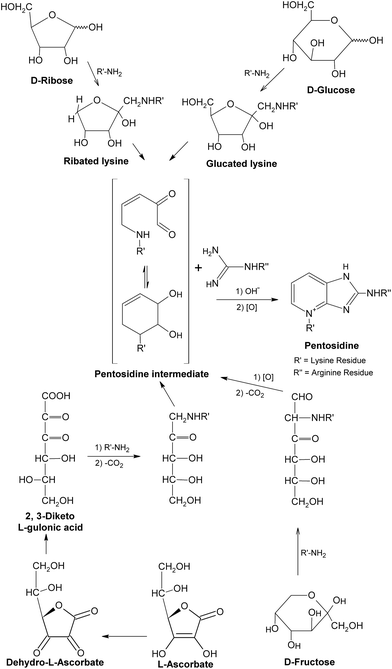 | ||
| Fig. 7 Formation of pentosidine. This figure was originally published in the Journal of Biological Chemistry. S. K. Grandhee and V. M. Monnier, J. Biol. Chem., 1991, 266, 11649–11653.57© the American Society for Biochemistry and Molecular Biology. | ||
As an example of non-fluorescent and non-crosslinking AGEs, CML is mainly thought to be formed by the oxidative breakdown of fructoselysine which is a product of glucose and lysine,62 as shown in Fig. 8. It was also indicated CML could be generated during the oxidation of ascorbic acid.63 Besides, the reactions between lysine and dicarbonyls formed in glycation (such as GO, MGO) would lead to the formation of CML as well.64 Possibly based on the above pathways, CML was formed in a variety of tissues, such as serum and urine of rats, and was considered as an index to assess the extent of glycoxidative damage with aging in rat kidney.65 Meanwhile, its levels in both skin collagen and serum of diabetic patients were reported to increase two-fold when compared to control (non-diabetic) patients, suggesting a potential link between CML and diabetes.34,66 Moreover, CML could also be formed under different food processing conditions, such as milk production and baking,67 and has become one of the most commonly used markers for the nutritional evaluation of heat-treated foods in terms of protein quality.68,69 When compared to some other AGEs, CML is more acid stable, facilitating its application as an indicator of the Maillard reaction progress both in biological and food systems.70
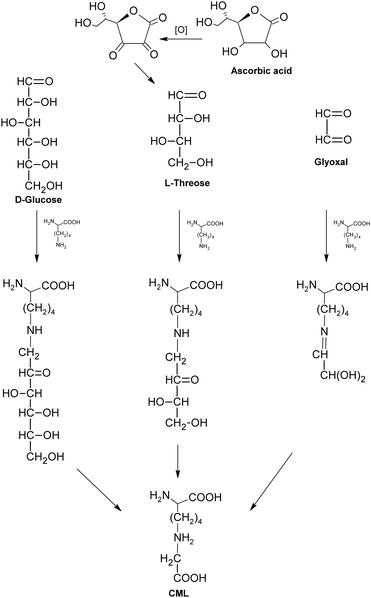 | ||
| Fig. 8 Formation of carboxymethyllysine (CML). | ||
During the early stage of glycation, some reactive dicarbonyl species including GO and MGO would be released as side-products of glycolysis (Fig. 9), leading to associated oxoaldehydes-derived AGE formation.71 Take MGO for an example. MGO has been proven to be one of the most reactive glycation agents,72,73 and could easily react with different groups of amino acids or proteins and form a series of MGO-derived AGEs.46 It has been reported that reactions between MGO and arginine residues may cause the formation of imidazolones (Fig. 10), such as Nδ-(5-hydro-5-methyl-4-imidazolon-2-yl)-L-ornithine (MG-H1), 2-amino-5-(2-amino-5-hydro-5-methyl-4-imidazolon-1-yl)pentanoic acid (MG-H2) and argpyrimidine,74–77 while reactions between MGO and lysine could yield MGO-derived lysine-lysine crosslinks (Fig. 10) Nε-(1-carboxyethyl)lysine (CEL) and MGO-derived lysine dimer (MOLD).78,79 In addition, reactions of MGO and the thiol group of cysteine would give rise to the formation of carboxyethylcysteine (Fig. 10), which was proposed for use as a marker in pathological states.80,81 These MGO derivatives were demonstrated to lead to the progression of diabetic complications, such as nephropathy and atherosclerosis,75,82 and contribute to the tissue injury, protein cross-linking and apoptosis.83
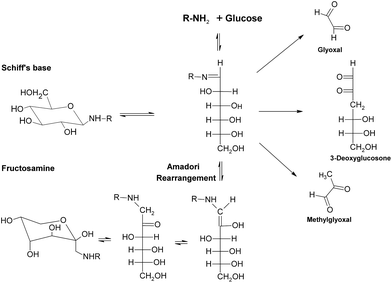 | ||
| Fig. 9 Formation of reactive dicarbonyls in glycation. | ||
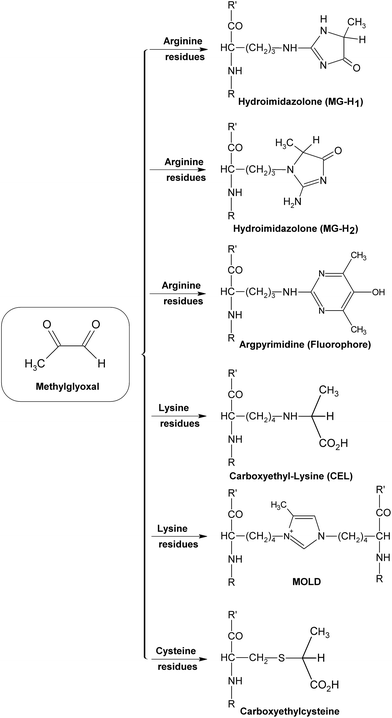 | ||
| Fig. 10 Formation of some methylglyoxal (MGO)-derived AGEs. | ||
On the other hand, MGO-induced chemical modification of proteins76 may have a significant impact on the functions of proteins. More and more evidence showed that the majority of the methylglyoxal present in vivo was bound to biological ligands84 and some research studies have pointed out that glycation caused by MGO could inactivate key cellular enzymes including glyceraldehyde-3-phophate dehydrogenase, glutathione reductase and lactate dehydrogenase,85 impair the activity of ribonuclease A,86 and lead to the loss of albumin antioxidant capacity.87 As proteins play important biological roles in both intracellular and extracellular activities, MGO-associated modification of proteins could also interfere with relevant cellular pathways and affect cellular functionality.88,89 Thus, effects of MGO on cellular functions have been widely investigated in different cell lines. Results showed that MGO is cytotoxic and could cause apoptosis in cultured human leukemia 60 (HL60) cells and decrease the viability of Chinese hamster ovary cells.90,91 Considering the detrimental effects of MGO, several MGO scavengers, such as aminoguanidine (AG) and tenilsetam, have been used as protectors to reduce negative impacts of MGO on proteins and cellular activities in cells. It was found that both AG and tenilsetam could decrease the toxic effects of MGO on human neuroblastoma cells.92 In rat L6 myoblasts, AG could hamper MGO-associated impairment of insulin signaling pathways to some extent.93 Another recognized MGO scavenger, N-acetyl cysteine, has also been demonstrated its ability to reserve both increased MGO level and insulin resistance state in 3T3-L1 adipocytes.94 These findings may offer favorable candidates for future drug development aiming at alleviating negative effects from MGO-associated disorders. Cell line models provide a relatively rapid, simple and direct platform for evaluating effects of extraneous stimulations (compounds) on cellular functions, facilitating the downstream animal studies or clinical trials, and become a useful tool in the fields of biochemistry and medicine.
3.3 Harmful effects of AGEs
The non-enzymatic glycation in vivo has been proved to be closely related to the pathogenesis of some chronic diseases, such as typical diabetic complications, atherosclerosis, Alzheimer's disease, rheumatoid arthritis and chronic heart failure,95–98 which have been mainly ascribed to the accumulation of AGEs. Meanwhile, reactive oxygen species (ROS) formed during the glycation process99 could cause oxidative stress and damage to tissues, which are also common end points of the above diseases.52,100Some studies have also indicated that the pathogenesis of rheumatoid arthritis is partially related to harmful changes induced by AGEs in the collagen network in bone which have been associated with a loss of toughness of bone in the elderly as well.118,119 Meanwhile, endothelial dysfunction in patients with chronic kidney disease has been proven to be mediated by AGE-induced inhibition of endothelial nitric oxide synthase via the activation of receptor for AGEs.120 Additionally, AGE-formed cross-liking could lead to vascular and myocardial stiffening, which are properties of the pathogenesis of chronic heart failure.95
3.4 Inhibition of the formation of AGEs
As the formation of AGEs in vivo has been involved in the pathogenesis of some chronic diseases, especially diabetic complications, the discovery and investigation of AGE inhibitors would offer a potential therapeutic approach for the prevention of diabetic or other pathogenic complications. So far, some potential AGE inhibitors have been proposed121,122 and several of them have reached the phase of clinical studies.15,123 Basically, AGE inhibitors can be divided into two groups: synthetic compounds and natural products. Generally speaking, the underlying inhibitory mechanisms of AGE inhibitors were mainly through the ways of blocking sugar attachment to proteins, attenuating glycoxidation and oxidative stress through trapping or scavenging some intermediates including reactive dicarbonyls, free radicals and nitrogen species produced in the process of glycation, and breaking down formed AGE crosslinks.124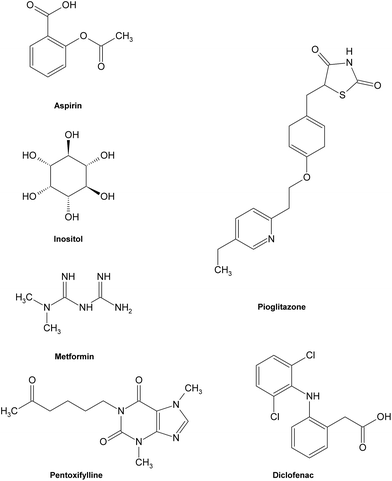 | ||
| Fig. 11 Structures of some typical AGE inhibitors effective in early glycation. | ||
Most synthetic inhibitors play a major role in preventing the formation of AGEs at the late stage of glycation (Fig. 12), which is mainly ascribed to their scavenging abilities on both reactive carbonyls and radicals formed during glycation, or blocking the formation of intermediate Amadori products. Aminoguanidine (AG) and pyridoxamine which are considered as potent carbonyl scavengers have been widely investigated as typical AGE inhibitors. AG, a nucleophilic hydrazine derivative, showed a strong ability to react with β-dicarbonyl intermidiates induced by glycation,134–136 and this activity is closely relevant to its prevention of the development of diabetic complications.14 When compared to AG,137 pyridoxamine was found to be more effective in inhibiting antigenic AGE formation in a bovine serum albumin model138–140 by trapping dicarbonyl compounds.139,141 Another dicarbonyl scavenger, thiamine pyrophosphate, also displayed a strong inhibitory effect on AGE formation which was comparable to that of pyridoxamine.142 Inhibitory effects on AGE formation of other carbonyl scavengers have also been studied. For example, tenilsetam was proved to restrain the polymerization of lysozyme with 3-DG in vitro and inhibited the formation of AGEs in the renal cortex and aorta.143 Metformin,144–146 buformin147 and carnosine148,149 all showed the potential to protect proteins against in vitro glycation and cross-linking and fought against the AGE-associated disorders in terms of their potency to reduce reactive carbonyls.150,151 Meanwhile, a series of other compounds, such as calcium antagonists,152 amlodipine,153 kinetin,154 quinine155 and synthesized 6-dimethylaminopyridoxamine,156 demonstrated their abilities to retard or suppress AGE formation possibly due to radical scavenging properties. There are also some anti-glycative compounds that exert their effects by inhibiting the formation of Amadori products. As an example, pyrraline could attach to sugar-derived moieties of glycated proteins, thus blocking the reactive sites for further polymerization reactions.92,157 Penicillamine was reported to decrease the formation of Amadori products158,159 and reduce levels of AGEs.160 Ethanol showed an obvious inhibitory effect on AGE formation as it could be metabolized to acetaldehyde in vivo that could react with protein-bound Amadori products.161
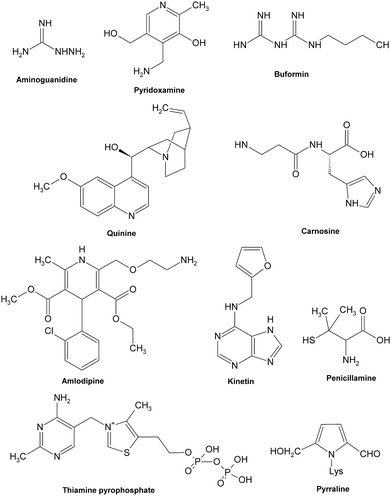 | ||
| Fig. 12 Structures of some typical AGE inhibitors effective in late glycation. | ||
Besides, a few cross-link breakers have also been investigated to combat deleterious AGE cross-links (Fig. 13), leading to the restoration of artery properties in experimental diabetes.162,163 Alagebrium chloride (ALT-711) was the first compound in the class of thiazolium that was reported to break down established AGE-related protein cross-links,164 which offers its ability to improve ventricular and arterial compliance,165 and ameliorate the adverse cardiovascular and renal changes associated with aging, diabetes, and hypertension.166–169 Another prototypic AGE cross-link breaker is N-phenacylthiazolium bromide (PTB)170 which can react with and cleave covalent AGE-derived protein cross-links. Other potent breakers such as curcumin171 and ALT-946172,173 also possess potential therapeutic value for diseases relative to AGE cross-links.
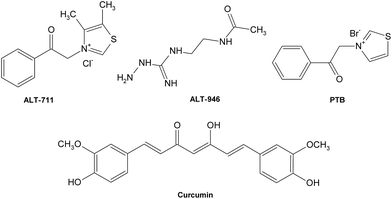 | ||
| Fig. 13 Structures of some typical AGE crosslink breakers. | ||
Although the synthetic compounds mentioned above have demonstrated strong power to inhibit the formation of AGEs or break cross-links, they might also cause severe side effects. Take AG for an example. AG, the first inhibitor engaging in clinical trials, had its ACTION II trial terminated due to safety concerns and inadequate efficacy. Relevant adverse effects observed in clinical trials of aminoguanidie including gastrointestinal disturbance, anemia, and flu-like symptoms13–15 cannot be ignored.
Extracts, fractions & compounds from medicinal plants. Extracts of two flavonoid-rich South American herbs, Achyrocline satureoides and Ilex paraguariensis, showed strong abilities to prevent the glycation of proteins induced by dicarbonyls at a 1/100 dilution of the herbal infusions, which were comparable to the inhibitory effects of millimolar amounts of aminoguanidine and carnosine as well as micromolar amounts of ascorbic acid.16 Besides, both antiglycation activity and antioxidant activity of aqueous ethanolic extracts from 25 plant tissues collected in Korea were determined and the few most effective ones in descending order were: Allium cepa (skin), Lllicium religiosum (bark and wood), Fagopyrum esculentum (hull), and Origanum officinalis (leaf). The results also suggested that the inhibitory activities against glycation were significantly correlated with the antioxidative capacities of the extracts.174 Meanwhile, aged garlic extract was suggested as a potential inhibitor for AGEs with potent antioxidant property.17,175 The studies of antioxidative, chelating, free radical scavenging and antiglycation activities of garcinol purified from the rind of Garcinia indica fruit (a garnish for curry and a traditional medicine in India) proved that garcinol might be a competent antioxidant and glycation inhibitor.18 The methanol extract from stems of Salacia chinensis (a medical foodstuff originating in Thailand) was found to possess a strong inhibitory effect on the formation of Amadori compounds and AGEs.176 From the leaves of Eucommia ulmoides O. (a folk remedy for the treatment of hypertention and diabetes used in Korea, Japan and China), a new flavonol glycoside, quercetin 3-O-α-L-arabinopyranosyl-β-D-glucopyranoside and known flavonols kaempferol 3-O-β-D-glucopyranoside and isoquercitrin were isolated. The flavonol glycosides showed antidiabetic action probably by inhibiting glycation.177 Aqueous extracts of Toona sinensis Roem. (Meliaceae) and Graptopetalum paragugayene E. Walther (Crassulaceae) were reported to prevent the formation of AGEs from low density lipoprotein (LDL) glycation induced by glucose and glyoxal.178 The inhibitory effects on AGE formation of some active components including isoflavone C-glucosides (such as puerarin and PG-3), isoflavone O-glucosides (such as daidzin and genistin) and pterocarpans (such as medicarpin, glycinol and tuberosin) and a new 2-arylbenzofuran (puerariafuran) isolated from the roots of Pueraria lobata were determined and isoflavone C-glucosides, isoflavone O-glucosides and puerariafuran showed stronger inhibitory activities than the positive control aminoguanidine.179,180 Besides, heat-processed ginseng (Panax ginseng C.A. Meyer) was reported to be capable of preventing renal damage and reduce the formation of AGEs in diabetic rats by alleviating oxidative stress.181 Some flavone C-glycosides were obtained from Zea mays L. (used in traditional Chinese medicine to treat dropsy and hypertension) and exhibited similar inhibitory effects as aminoguanidine.182–184 From the BuOH-soluble extract of the seeds of Cassia tora, three naphthopyrone glucosides, cassiaside, rubrofusarin-6-O-β-D-gentiobioside and toralactone-9-O-β-D-gentiobioside, were isolated as active components. All the isolates displayed inhibitory effects on the formation of AGEs in vitro.19 Two dihydroflavonol glycosides, engeletin and astilbin, were isolated from an EtOAc extract of the leaves of Stelechocarpus cauliflorus R.E. Fr. (Annonaceae), whose root is used for the treatment of stomach aches. Astilbin was more potent than engeletin to prevent the formation of AGEs, and was about as potent as a reported natural inhibitor quercetin.185 Furthermore, five crude drug constituents of Wen-Pi-Tang (Rhei Rhizoma, Ginseng Radix, Aconiti Tuber, Zingiberis Rhizoma and Glycyrrhizae Radix), a Chinese medical prescription to treat moderate renal failure, were chosen to examine their capability to prevent protein glycation. Rhei Rhizoma displayed the most potent activity, Zingiberis Rhizoma and Glycyrrhizae Radix showed relatively moderate activity and Aconiti Tuber and Ginseng Radix presented weak activity. Meanwhile, compounds like tannins, especially rhatannin, RG-tannin and procyanidin B-2 3,3′-di-O-gallate obtained from Rhei Rhizoma and Glycyrrhizae Radix exerted superior activities that were stronger than the positive control aminoguanidine. The isolated flavones such as licochalcone A and licochalcone B, and anthraquinones such as emodin and aloe-emodin, also showed obvious inhibitory activity against glycation.186,187 Additionally, some prenylated flavonoids isolated from Sophora flavescens, a traditional herbal medicine, possessed potent inhibitory effects on AGE formation when compared to the positive control aminoguanidine.188 Butein and sulfuretin obtained from Rhus verniciflua, which has various biological activities including anti-inflammaroty and anti-cancer, were also reported to have significant inhibitory effects on the formation of AGEs.189
Vegetables & fruits. It has been reported that the EtOAc extract of mustard leaf not only showed potent antiglycation activity and inhibitory effect on free radical-mediated protein damage in vitro, but also reduced the increased levels of superoxide and nitrite/nitrate, and protected against diabetic oxidative stress induced by streptozotocin in vivo.190 In addition, tomato paste strongly suppressed AGE formation chiefly due to one of its antioxidant components, rutin.191 From acerola fruit (Malpighia emarginata DC.), two anthocyanins and quercitrin were isolated and the radical scavenging and AGE formation inhibitory activities of these compounds were evaluated. All the three polyphenols demonstrated effective inhibition of AGE formation.192Psidium guajava L. is a tropical fruit and the leaves of which are used as a folk therapeutic in the treatment of diabetes and enteritis and its aqueous extract was demonstrated a potent antiglycative agent.178 Moreover, the polysaccharide fraction from Punica granatum (Pomegranate) was found to have high antioxidant activity, free radical scavenging activity and antiglycation activity.193
Teas. Green tea extract could reduce age-related increase in collagen cross-linking and fluorescent products in C57BL/6 mice194 and protect against protein oxidation and glycation mainly due to tannin components whose chemical structures were involved in the protective activity.195 In addition, the inhibitory effect of green tea extract was compared with Ilex paraguariensis (IP) extract. Neither of them presented significant participation as inhibitors in the early phase of the glycation process based on the results from the SDS-PAGE. IP extract mainly inhibited the free-radical mediated conversion of Amadori products to AGEs with activity comparable to that of aminoguanidine, whereas green tea extract was weaker.196 The aqueous extract from Luobuma tea was studied by the measurement of fluorescence AGEs formed in bovine serum saline with the addition of glucose. It showed obvious activity against AGE formation. Following further fractionations of the extract, seven polyphenolic compounds were isolated which presented more potent inhibitory effects against AGE formation than aminoguanidine.197
Cereals, spices, nuts & algae. The methanolic extracts of Finger millet (Eleusine coracana) and Kodo millet (Paspalum scrobiculatum) were found to be useful for protection from glycation and cross-linking of collagen.198 Among extracts of 34 spices, the methanol extract of thyme (Thymus vulgaris) showed the strongest activity in the inhibition of glycation of bovine serum albumin (BSA). Four flavonoids (quercetin, eriodictyol, 5,6,4′-trihydroxy-7,8,3′-trimethoxyflavone and cirsilineol) were isolated from thyme by chromatographic purification. They all lowered the levels of AGE formation.199 Eight flavonoids isolated from the water-soluble fraction of peanut skin were evaluated for their free radical scavenging activity and protein glycation inhibitory effects.200 From the brown alga, Eisenia bicyclis, a new phloroglucinol derivative, eckol and dieckol were isolated and proven to possess obvious inhibitory activity against glycation.201
Purified phenolic compounds. It is noted that the inhibitory effects on the formation of AGEs of extracts from plants are largely contributed by the abundant amount of polyphenolics they contain. Polyphenolics including flavonoids are well known antioxidants and they may trap the oxidative free radicals generated during the process of AGE formation to thus decrease the final levels of AGEs. Hence, some studies have directly focused on the inhibitory activities of some naturally occurring polyphenolics and their derivatives.
G-rutin, a water soluble rutin glucose derivative, was tested for its in vitro antioxidant activity and tissue anti-glycative property in three body protein sources susceptible to initial and advanced glycation reactions. The findings indicated that G-rutin is a potent glycation inhibitor, especially for kidney proteins.202 Moreover, rutin and its five circulating metabolites could inhibit both fluorescent and nonfluorescent AGEs during the glycation of collagen induced by glucose in vitro. Among its metabolites, those containing vicinyl dihydroxyl groups had stronger inhibitory effects on the formation of some AGEs including pentosidine and fluorescent adducts than metabolites without vicinyl dihydroxyl groups. All the five metabolites of rutin were effective to prevent the formation of Nε-carboxymethyllysine (CML) adducts.203
In another experiment, the antiglycative effects of some plant phenolics were investigated in an in vitro system composed of porcine aspartate aminotransferase (AST) and fructose. Of these natural antioxidants, baicalin and baicalein which are flavones and quinic acid had no antiglycation effect. Others like arbutin, ferulic acid and hydroxycitric acid showed obvious inhibitory action of glycation.204
Ten naturally occurring flavonoids were also studied on the prevention of hyperglycemia-mediated protein modification at the individual stages of protein glycation in vitro. Therein, luteolin had a pronounced inhibitory effect at each stage of protein glycation, and other flavonoids all displayed effective antiglycation activities to different extents. Besides, a strong and positive correlation was demonstrated between inhibitory effects and free radical scavenging activities.205 In general, the flavonoids with evident inhibitory activities also possess strong radical scavenging activities.206 Moreover, the decrease in glycation by flavonoids was associated with an increase in the content of antioxidant components determined by the levels and activities of thiol-containing proteins including glutathione peroxidase.207
Structure–activity relationships of flavonoids with antiglycative effects. In general, flavones exhibit stronger inhibitory effects on the formation of AGEs when compared to flavonols, flavanones, and isoflavones (Fig. 14), with several exceptions.204 The inhibitory effects were also in the order of flavonol > flavanol > flavanone.205 Furthermore, anthocyanins displayed more potent inhibitory abilities than their corresponding glycosides.
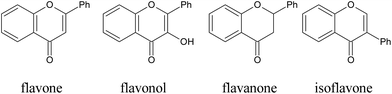 | ||
| Fig. 14 Structures of some typical flavonoids. | ||
When it comes to the functional groups, for flavonoids, the hydroxyl group at the C-3′ position is beneficial to inhibitory activity in AGE formation while the hydroxyl group at the C-3 position is not an essential requirement.205 The single hydroxyl group at 5′-position of the B ring likely contributes to the inhibitory activity.190,200 When the number of hydroxyl groups increases at positions including 3′-, 4′-, 5-, and 7, the inhibitory activity was enhanced correspondingly. Besides, methylation and glycosylation of the hydroxyl groups at certain positions also influence the inhibitory activity in glycation. For example, methylation of the 3-OH group of flavonols led to increased inhibitory activity, whereas glycosylation of the hydroxyl group at the C-7 position, and methylation or glycosylation of the hydroxyl group at 4′-hydroxyl group of flavones, flavonols and flavanones appeared to decrease the activity.200,206 However, all these structure–activity relationships are only based on an in vitro study. In an in vivo study, totally different results may be obtained.
Potential inhibitory mechanisms of flavonoids. So far, the inhibitory effects of phenolic compounds on AGE formation are mainly thought to be contributed by their potent antioxidant activities, which provide them strong abilities to scavenge free radicals formed during glycation, thus influence the subsequent formation of AGEs. However, limited effort has been devoted to understanding the underlying mechanisms of effective natural AGE inhibitors. Recently, it has been reported that several tea polyphenols (Fig. 15) possess abilities to scavenge certain intermediate RCS during glycation under physiological conditions,208 thus blocking subsequent pathyways in AGE formation, which suggests the antioxidant property might not be the only reason for phenolic compounds to inhibit AGE formation. On the other hand, natural AGE inhibitors with both antioxidant activities and carbonyl scavenging capacities may not only alleviate AGE-caused health disorders, but also prevent from RCS-induced detrimental symptoms.
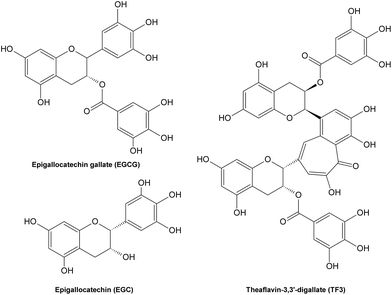 | ||
| Fig. 15 Structures of some tea polyphenols with carbonyl scavenging abilities. | ||
4 Summary
Increased formation and accumulation of AGEs have been implicated in the pathogenesis of diabetic complications and other relevant diseases. Recent studies of inhibition against AGE formation have led to the development of various AGE inhibitors and AGE-cross-link breakers both from synthetic compounds and natural products. However, synthetic AGE inhibitors might not be suitable to be further developed as drugs since they may induce adverse effects during clinical trials. Moreover, the complicated process for their synthesis is time-consuming and expensive. For this consideration, more and more attention has been paid to plant-derived polyphenolic compounds with a hope to identify potent antiglycative agents. The antiglycative activities of natural inhibitors were proven to be concentration-dependent and have a positive relationship with their respective antioxidant activities in most cases. For certain natural AGE inhibitors, abilities to scavenge reactive intermediate carbonyls during glycation also contribute to their suppression of AGE formation. To sum up, AGE inhibitors from natural products with relatively low toxicity are more promising candidates for the development of functional additives, nutraceuticals and even drugs for the treatment of diabetic complications and other AGE-asscociated diseases.References
- R. Singh, A. Barden, T. Mori and L. Beilin, Diabetologia, 2001, 44, 129–146 Search PubMed.
- S. E. Fayle and J. A. Gerrard, in The Maillard Reaction, ed. P. S. Belton, Royal Society of Chemistry, Cambridge, 2002, ch. 1, pp. 1–6 Search PubMed.
- H. E. Nursten, in The Maillard Reaction: Chemistry, Biochemistry, and Implications, ed. H. Nursten, Royal Society of Chemistry, Cambridge, 2005. ch. 1, pp.1–4 Search PubMed.
- S. Rahbar, Clin. Chim. Acta, 1968, 20, 381–385 Search PubMed.
- S. Rahbar, O. Blumenfeld and H. M. Ranney, Biochem. Biophys. Res. Commun., 1969, 36, 838–843 Search PubMed.
- N. Ahmed, Diabetes Res. Clin. Pract., 2005, 67, 3–21 Search PubMed.
- T. Koschinsky, C. J. He, T. Mitsuhashi, R. Bucala, C. Liu, C. Buenting, K. Heitmann and H. Vlassara, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6474–6479 Search PubMed.
- M. Brownlee, Diabetes, 1994, 43, 836–841 Search PubMed.
- S. Kume, M. Takeya, T. Mori, N. Araki, H. Suzuki, S. Horiuchi, T. Kodama, Y. Miyauchi and K. Takahashi, Am. J. Pathol., 1995, 147, 654–667 Search PubMed.
- M. P. Vitek, K. Bhattacharya, J. M. Glendening, E. Stopa, H. Vlassara, R. Bucala, K. Manogue and A. Cerami, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4766–4770 Search PubMed.
- G. Munch, J. Thome, P. Foley, R. Schinzel and P. Riederer, Brain Res. Rev., 1997, 23, 134–143 Search PubMed.
- M. Brownlee, Annu. Rev. Med., 1995, 46, 223–234 CrossRef CAS.
- B. I. Freedman, J. P. Wuerth, K. Cartwright, R. P. Bain, S. Dippe, K. Hershon, A. D. Mooradian and B. S. Spinowitz, Controlled Clin. Trials, 1999, 20, 493–510 Search PubMed.
- P. J. Thornalley, Arch. Biochem. Biophys., 2003, 419, 31–40 Search PubMed.
- M. E. Williams, Curr. Diabetes Rep., 2004, 4, 441–446 Search PubMed.
- A. Gugliucci and T. Menini, Life Sci., 2002, 72, 279–292 Search PubMed.
- J. Imai, N. Ide, S. Nagae, T. Moriguchi, H. Matsuura and Y. Itakura, Planta Med., 1994, 60, 417–420 CrossRef CAS.
- F. Yamaguchi, T. Ariga, Y. Yoshimura and H. Nakazawa, J. Agric. Food Chem., 2000, 48, 180–185 CrossRef CAS.
- G. Y. Lee, D. S. Jang, Y. M. Lee, J. M. Kim and J. S. Kim, Arch. Pharmacal Res., 2006, 29, 587–590 Search PubMed.
- L. Maillard, Comptes Rendus Hebdomadaires des Seances de l Academie des Sciences, 1912, 154, 66–68 Search PubMed.
- G. Suarez, R. Rajaram, A. L. Oronsky and M. A. Gawinowicz, J. Biol. Chem., 1989, 264, 3674–3679 Search PubMed.
- R. Bucala, P. Model and A. Cerami, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 105–109 CAS.
- H. S. Isbell and H. L. Frush, J. Org. Chem., 1958, 23, 1309–1319 CrossRef CAS.
- F. G. Njoroge and V. M. Monnier, Prog. Clin. Biol. Res., 1989, 304, 85–107 Search PubMed.
- C. Delgado-Andrade and F. J. Morales, J. Agric. Food Chem., 2005, 53, 1403–1407 CrossRef CAS.
- M. J. Lane and H. E. Nursten, in The Maillard Reaction in Food and Nutrition, ed. G. R. Waller and M. S. Feather, American Chemical Society, 1983, vol. 215, ch. 9, pp. 141–158 Search PubMed.
- M. E. Bruins, A. J. H. Thewessen, A. E. M. Janssen and R. M. Boom, J. Mol. Catal. B: Enzym., 2003, 21, 31–34 CrossRef CAS.
- E. Tareke, P. Rydberg, P. Karlsson, S. Eriksson and M. Tornqvist, J. Agric. Food Chem., 2002, 50, 4998–5006 CrossRef CAS.
- G. A. Gross, R. J. Turesky, L. B. Fay, W. G. Stillwell, P. L. Skipper and S. R. Tannenbaum, Carcinogenesis, 1993, 14, 2313–2318 Search PubMed.
- N. L. Tran, C. P. Salmon, M. G. Knize and M. E. Colvin, Food Chem. Toxicol., 2002, 40, 673–684 Search PubMed.
- S. Rahbar and J. Figarola, Curr. Med. Chem.: Immunol., Endocr. Metab. Agents, 2002, 2, 135–161 Search PubMed.
- K. Ikeda, T. Higashi, H. Sano, Y. Jinnouchi, M. Yoshida, T. Araki, S. Ueda and S. Horiuchi, Biochemistry, 1996, 35, 8075–8083 CrossRef CAS.
- M. A. Smith, S. Taneda, P. L. Richey, S. Miyata, S. D. Yan, D. Stern, L. M. Sayre, V. M. Monnier and G. Perry, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5710–5714 CrossRef CAS.
- D. G. Dyer, J. A. Dunn, S. R. Thorpe, K. E. Bailie, T. J. Lyons, D. R. McCance and J. W. Baynes, J. Clin. Invest., 1993, 91, 2463–2469 CrossRef CAS.
- D. R. McCance, D. G. Dyer, J. A. Dunn, K. E. Bailie, S. R. Thorpe, J. W. Baynes and T. J. Lyons, J. Clin. Invest., 1993, 91, 2470–2478 Search PubMed.
- E. B. Frye, T. P. Degenhardt, S. R. Thorpe and J. W. Baynes, J. Biol. Chem., 1998, 273, 18714–18719 Search PubMed.
- T. Henle, U. Schwarzenbolz and H. Klostermeyer, Eur. Food Res. Technol., 1997, 204, 95–98 Search PubMed.
- N. Ahmed, B. Mirshekar-Syahkal, L. Kennish, N. Karachalias, R. Babaei-Jadidi and P. J. Thornalley, Mol. Nutr. Food Res., 2005, 49, 691–699 Search PubMed.
- T. Henle, Kidney Int., 2003, S145–147 Search PubMed.
- T. Goldberg, W. Cai, M. Peppa, V. Dardaine, B. S. Baliga, J. Uribarri and H. Vlassara, J. Am. Diet. Assoc., 2004, 104, 1287–1291 Search PubMed.
- M. X. Fu, K. J. Wells-Knecht, J. A. Blackledge, T. J. Lyons, S. R. Thorpe and J. W. Baynes, Diabetes, 1994, 43, 676–683 CrossRef CAS.
- A. M. Schmidt, O. Hori, J. Brett, S. D. Yan, J. L. Wautier and D. Stern, Arterioscler. Thromb., 1994, 14, 1521–1528 Search PubMed.
- A. M. Schmidt, S. D. Yan, J. L. Wautier and D. Stern, Circ. Res., 1999, 84, 489–497 CAS.
- M. Beranek, D. Novakova, P. Rozsival, J. Drsata and V. Palicka, Acta Medica (Hradec Kralove), 2006, 49, 35–39 Search PubMed.
- A. Bierhaus, M. A. Hofmann, R. Ziegler and P. P. Nawroth, Cardiovasc. Res., 1998, 37, 586–600 Search PubMed.
- T. W. Lo, M. E. Westwood, A. C. McLellan, T. Selwood and P. J. Thornalley, J. Biol. Chem., 1994, 269, 32299–32305 CAS.
- M. X. Fu, J. R. Requena, A. J. Jenkins, T. J. Lyons, J. W. Baynes and S. R. Thorpe, J. Biol. Chem., 1996, 271, 9982–9986 Search PubMed.
- S. P. Wolff and R. T. Dean, Biochem. J., 1987, 245, 243–250 CAS.
- S. P. Wolff, Z. Y. Jiang and J. V. Hunt, Free Radical Biol. Med., 1991, 10, 339–352 CrossRef CAS.
- T. Miyata, C. van Ypersele de Strihou, K. Kurokawa and J. W. Baynes, Kidney Int., 1999, 55, 389–399 Search PubMed.
- T. Miyata, Bull. Mem. Acad. R. Med. Belg., 2002, 157, 189–196 Search PubMed.
- J. W. Baynes and S. R. Thorpe, Diabetes, 1999, 48, 1–9 Search PubMed.
- A. Gasser and J. M. Forbes, Protein Pept. Lett., 2008, 15, 385–391 Search PubMed.
- K. M. Biemel, O. Reihl, J. Conrad and M. O. Lederer, J. Biol. Chem., 2001, 276, 23405–23412 Search PubMed.
- P. Chellan and R. H. Nagaraj, J. Biol. Chem., 2001, 276, 3895–3903 Search PubMed.
- D. G. Dyer, J. A. Blackledge, S. R. Thorpe and J. W. Baynes, J. Biol. Chem., 1991, 266, 11654–11660 CAS.
- S. K. Grandhee and V. M. Monnier, J. Biol. Chem., 1991, 266, 11649–11653 CAS.
- A. Pillin, F. Pudil, V. Bencko and D. Bezdickova, Soudni lekarstvi/casopis Sekce soudniho lekarstvi Cs. lekarske spolecnosti J. Ev, 2007, 52, 60–64 Search PubMed.
- D. R. Sell and V. M. Monnier, J. Clin. Invest., 1990, 85, 380–384 Search PubMed.
- D. R. Sell, A. Lapolla, P. Odetti, J. Fogarty and V. M. Monnier, Diabetes, 1992, 41, 1286–1292 CrossRef CAS.
- D. R. Sell, R. H. Nagaraj, S. K. Grandhee, P. Odetti, A. Lapolla, J. Fogarty and V. M. Monnier, Diabetes/Metab. Rev., 1991, 7, 239–251 Search PubMed.
- M. U. Ahmed, S. R. Thorpe and J. W. Baynes, J. Biol. Chem., 1986, 261, 4889–4894 Search PubMed.
- J. A. Dunn, M. U. Ahmed, M. H. Murtiashaw, J. M. Richardson, M. D. Walla, S. R. Thorpe and J. W. Baynes, Biochemistry, 1990, 29, 10964–10970 Search PubMed.
- Y. Al-Abed and R. Bucala, Bioorg. Med. Chem. Lett., 1995, 5, 2161–2162 Search PubMed.
- M. Hamelin, C. Borot-Laloi, B. Friguet and H. Bakala, Arch. Biochem. Biophys., 2003, 411, 215–222 Search PubMed.
- J. Miura, S. Yamagishi, Y. Uchigata, M. Takeuchi, H. Yamamoto, Z. Makita and Y. Iwamoto, J. Diabetes Complications, 2003, 17, 16–21 Search PubMed.
- J. M. Ames, Ann. N. Y. Acad. Sci., 2008, 1126, 20–24 Search PubMed.
- J. Hartkopf and H. F. Erbersdobler, Z. Lebensm.-Unters. Forsch., 1994, 198, 15–19 Search PubMed.
- S. Drusch, V. Faist and H. F. Erbersdobler, Food Chem., 1999, 65, 547–553 CrossRef CAS.
- S. H. Assar, C. Moloney, M. Lima, R. Magee and J. M. Ames, Amino Acids, 2009, 36, 317–326 Search PubMed.
- P. J. Thornalley, A. Langborg and H. S. Minhas, Biochem. J., 1999, 344 Pt 1, 109–116 Search PubMed.
- M. Shinohara, P. J. Thornalley, I. Giardino, P. Beisswenger, S. R. Thorpe, J. Onorato and M. Brownlee, J. Clin. Invest., 1998, 101, 1142–1147 CrossRef CAS.
- S. J. Meade, A. G. Miller and J. A. Gerrard, Bioorg. Med. Chem., 2003, 11, 853–862 CrossRef CAS.
- M. E. Westwood and P. J. Thornalley, J. Protein Chem., 1995, 14, 359–372 Search PubMed.
- T. Oya, N. Hattori, Y. Mizuno, S. Miyata, S. Maeda, T. Osawa and K. Uchida, J. Biol. Chem., 1999, 274, 18492–18502 CrossRef CAS.
- T. P. Degenhardt, S. R. Thorpe and J. W. Baynes, Cell Mol. Biol. (Noisy-le-grand), 1998, 44, 1139–1145 Search PubMed.
- Y. Nakadate, K. Uchida, K. Shikata, S. Yoshimura, M. Azuma, T. Hirata, H. Konishi, H. Kiyama and T. Tachibana, Biochem. Biophys. Res. Commun., 2009, 378, 209–212 Search PubMed.
- H. Odani, T. Shinzato, J. Usami, Y. Matsumoto, E. Brinkmann Frye, J. W. Baynes and K. Maeda, FEBS Lett., 1998, 427, 381–385 Search PubMed.
- M. U. Ahmed, E. Brinkmann Frye, T. P. Degenhardt, S. R. Thorpe and J. W. Baynes, Biochem. J., 1997, 324(Pt 2), 565–570 Search PubMed.
- J. Zeng and M. J. Davies, Chem. Res. Toxicol., 2005, 18, 1232–1241 Search PubMed.
- J. M. Acimovic, B. D. Stanimirovic and L. M. Mandic, J. Serb. Chem. Soc., 2009, 74, 867–883 Search PubMed.
- M. Bourajjaj, C. D. Stehouwer, V. W. van Hinsbergh and C. G. Schalkwijk, Biochem. Soc. Trans., 2003, 31, 1400–1402 Search PubMed.
- R. Ramasamy, S. F. Yan and A. M. Schmidt, Cell, 2006, 124, 258–260 Search PubMed.
- F. W. Chaplen, W. E. Fahl and D. C. Cameron, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5533–5538 Search PubMed.
- P. E. Morgan, R. T. Dean and M. J. Davies, Arch. Biochem. Biophys., 2002, 403, 259–269 Search PubMed.
- A. G. Miller, S. J. Meade and J. A. Gerrard, Bioorg. Med. Chem., 2003, 11, 843–852 Search PubMed.
- P. Faure, L. Troncy, M. Lecomte, N. Wiernsperger, M. Lagarde, D. Ruggiero and S. Halimi, Diabetes Metab., 2005, 31, 169–177 Search PubMed.
- D. Yao, T. Taguchi, T. Matsumura, R. Pestell, D. Edelstein, I. Giardino, G. Suske, N. Ahmed, P. J. Thornalley, V. P. Sarthy, H. P. Hammes and M. Brownlee, Cell, 2006, 124, 275–286 CrossRef CAS.
- J. H. Kang, Journal of Biochemistry and molecular biology, 2006, 39, 335–338 Search PubMed.
- P. J. Thornalley, Gen. Pharmacol., 1996, 27, 565–573 Search PubMed.
- F. W. Chaplen, Cytotechnology, 1998, 26, 173–183 Search PubMed.
- J. Webster, C. Urban, K. Berbaum, C. Loske, A. Alpar, U. Gartner, S. G. de Arriba, T. Arendt and G. Munch, Neurotoxic. Res., 2005, 7, 95–101 Search PubMed.
- A. Riboulet-Chavey, A. Pierron, I. Durand, J. Murdaca, J. Giudicelli and E. Van Obberghen, Diabetes, 2006, 55, 1289–1299 Search PubMed.
- X. Jia and L. Wu, Mol. Cell. Biochem., 2007, 306, 133–139 Search PubMed.
- A. J. Smit, J. W. Hartog, A. A. Voors and D. J. van Veldhuisen, Ann. N. Y. Acad. Sci., 2008, 1126, 225–230 Search PubMed.
- R. Meerwaldt, T. Links, C. Zeebregts, R. Tio, J.-L. Hillebrands and A. Smit, Cardiovascular Diabetology, 2008, 7, 29 Search PubMed.
- A. J. Smit and H. L. Lutgers, Curr. Med. Chem., 2004, 11, 2767–2784 Search PubMed.
- H. Vlassara and M. R. Palace, J. Intern. Med., 2002, 251, 87–101 CrossRef CAS.
- T. Nishikawa, D. Edelstein, X. L. Du, S. Yamagishi, T. Matsumura, Y. Kaneda, M. A. Yorek, D. Beebe, P. J. Oates, H. P. Hammes, I. Giardino and M. Brownlee, Nature, 2000, 404, 787–790 Search PubMed.
- T. Miyata, N. Ishikawa and C. van Ypersele de Strihou, Clin. Chem. Lab. Med., 2003, 41, 1150–1158 Search PubMed.
- M. Peppa and H. Vlassara, Hormones (Athens), 2005, 4, 28–37 Search PubMed.
- A. W. Stitt, A. J. Jenkins and M. E. Cooper, Expert Opin. Invest. Drugs, 2002, 11, 1205–1223 Search PubMed.
- M. Brownlee, Nature, 2001, 414, 813–820 CrossRef CAS.
- M. Brownlee, Diabetes, 2005, 54, 1615–1625 CrossRef CAS.
- A. W. Stitt, Exp. Mol. Pathol., 2003, 75, 95–108 Search PubMed.
- S. Uhlmann, K. Rezzoug, U. Friedrichs, S. Hoffmann and P. Wiedemann, Graefe's Arch. Clin. Exp. Ophthalmol., 2002, 240, 860–866 Search PubMed.
- R. Chibber, P. A. Molinatti, N. Rosatto, B. Lambourne and E. M. Kohner, Diabetologia, 1997, 40, 156–164 Search PubMed.
- A. Stevens, J. Am. Optom. Assoc., 1998, 69, 519–530 Search PubMed.
- V. M. Monnier, D. R. Sell, R. H. Nagaraj, S. Miyata, S. Grandhee, P. Odetti and S. A. Ibrahim, Diabetes, 1992, 41(Suppl. 2), 36–41 Search PubMed.
- H. Vlassara, M. Brownlee and A. Cerami, Diabetes, 1985, 34, 553–557 Search PubMed.
- M. Brownlee, H. Vlassara and A. Cerami, Diabetes, 1986, 35, 999–1003 Search PubMed.
- A. Gugliucci and M. Bendayan, Biochem. Biophys. Res. Commun., 1995, 212, 56–62 CrossRef CAS.
- H. Vlassara, Ann. Med., 1996, 28, 419–426 Search PubMed.
- C. W. Yang, H. Vlassara, E. P. Peten, C. J. He, G. E. Striker and L. J. Striker, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 9436–9440 Search PubMed.
- M. E. Westwood and P. J. Thornalley, Immunol. Lett., 1996, 50, 17–21 Search PubMed.
- V. V. Shuvaev, I. Laffont, J. M. Serot, J. Fujii, N. Taniguchi and G. Siest, Neurobiol. Aging, 2001, 22, 397–402 Search PubMed.
- C. A. Colaco, M. D. Ledesma, C. R. Harrington and J. Avila, Nephrol. Dial. Transplant, 1996, 11(Suppl. 5), 7–12 Search PubMed.
- J. DeGroot, N. Verzijl, M. J. Wenting-Van Wijk, R. A. Bank, F. P. Lafeber, J. W. Bijlsma and J. M. TeKoppele, Arthritis Rheum., 2001, 44, 2562–2571 Search PubMed.
- X. Wang, X. Shen, X. Li and C. M. Agrawal, Bone, 2002, 31, 1–7 CrossRef.
- E. Linden, W. Cai, J. C. He, C. Xue, Z. Li, J. Winston, H. Vlassara and J. Uribarri, Clin. J. Am. Soc. Nephrol., 2008, 3, 691–698 Search PubMed.
- S. Rahbar and J. L. Figarola, Arch. Biochem. Biophys., 2003, 419, 63–79 Search PubMed.
- S. Rahbar, K. K. Yerneni, S. Scott, N. Gonzales and I. Lalezari, Mol. Cell Biol. Res. Commun., 2000, 3, 360–366 Search PubMed.
- N. Giannoukakis, Curr. Opin. Investig. Drugs, 2005, 6, 410–418 Search PubMed.
- V. P. Reddy and A. Beyaz, Drug Discovery Today, 2006, 11, 646–654 Search PubMed.
- N. S. Malik and K. M. Meek, Biochem. Biophys. Res. Commun., 1994, 199, 683–686 Search PubMed.
- F. Caballero, E. Gerez, A. Batlle and E. Vazquez, Chem.-Biol. Interact., 2000, 126, 215–225 Search PubMed.
- G. V. Shastri, M. Thomas, A. J. Victoria, R. Selvakumar, A. S. Kanagasabapathy, K. Thomas and Lakshmi, Indian J. Exp. Biol., 1998, 36, 651–657 Search PubMed.
- M. A. van Boekel, P. J. van den Bergh and H. J. Hoenders, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1992, 1120, 201–204 Search PubMed.
- S. Ramakrishnan, K. N. Sulochana and R. Punitham, Indian J. Biochem. Biophys., 1999, 36, 129–133 Search PubMed.
- E. J. Menzel and R. Reihsner, Ann. Clin. Biochem., 1996, 33(Pt 3), 241–248 Search PubMed.
- E. J. Menzel and R. Reihsner, Diabetologia, 1991, 34, 12–16 Search PubMed.
- J. D. Mendez and L. I. Leal, Biomed. Pharmacother., 2004, 58, 598–604 Search PubMed.
- S. Rahbar, R. Natarajan, K. Yerneni, S. Scott, N. Gonzales and J. L. Nadler, Clin. Chim. Acta, 2000, 301, 65–77 Search PubMed.
- M. Brownlee, H. Vlassara, A. Kooney, P. Ulrich and A. Cerami, Science, 1986, 232, 1629–1632 CAS.
- J. A. Corbett, R. G. Tilton, K. Chang, K. S. Hasan, Y. Ido, J. L. Wang, M. A. Sweetland, J. R. Lancaster, Jr., J. R. Williamson and M. L. McDaniel, Diabetes, 1992, 41, 552–556 Search PubMed.
- S. Picard, S. Parthasarathy, J. Fruebis and J. L. Witztum, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 6876–6880 CrossRef CAS.
- A. A. Booth, R. G. Khalifah and B. G. Hudson, Biochem. Biophys. Res. Commun., 1996, 220, 113–119 CrossRef CAS.
- T. O. Metz, N. L. Alderson, S. R. Thorpe and J. W. Baynes, Arch. Biochem. Biophys., 2003, 419, 41–49 Search PubMed.
- P. A. Voziyan, T. O. Metz, J. W. Baynes and B. G. Hudson, J. Biol. Chem., 2002, 277, 3397–3403 Search PubMed.
- R. G. Khalifah, J. W. Baynes and B. G. Hudson, Biochem. Biophys. Res. Commun., 1999, 257, 251–258 CrossRef CAS.
- N. Ahmed, R. Luthen, D. Haussinger, K. Sebekova, R. Schinzel, W. Voelker, A. Heidland and P. J. Thornalley, Ann. N. Y. Acad. Sci., 2005, 1043, 718–724 Search PubMed.
- A. A. Booth, R. G. Khalifah, P. Todd and B. G. Hudson, J. Biol. Chem., 1997, 272, 5430–5437 Search PubMed.
- H. Shoda, S. Miyata, B. F. Liu, H. Yamada, T. Ohara, K. Suzuki, M. Oimomi and M. Kasuga, Endocrinology, 1997, 138, 1886–1892 Search PubMed.
- D. Ruggiero-Lopez, M. Lecomte, G. Moinet, G. Patereau, M. Lagarde and N. Wiernsperger, Biochem. Pharmacol., 1999, 58, 1765–1773 Search PubMed.
- P. Beisswenger and D. Ruggiero-Lopez, Diabetes Metab., 2003, 29, 95–103 Search PubMed.
- P. J. Beisswenger, S. K. Howell, A. D. Touchette, S. Lal and B. S. Szwergold, Diabetes, 1999, 48, 198–202 Search PubMed.
- T. Kiho, M. Kato, S. Usui and K. Hirano, Clin. Chim. Acta, 2005, 358, 139–145 Search PubMed.
- A. R. Hipkiss, J. Michaelis, P. Syrris, S. Kumar and Y. Lam, Biochem. Soc. Trans., 1994, 22, 399 Search PubMed.
- H. Yan and J. J. Harding, Biochim. Biophys. Acta., 2005, 1741, 120–126 Search PubMed.
- D. Bonnefont-Rousselot, J. Soc. Biol., 2001, 195, 391–398 Search PubMed.
- A. R. Hipkiss, Int. J. Biochem. Cell Biol., 1998, 30, 863–868 CrossRef CAS.
- G. Sobal, E. J. Menzel and H. Sinzinger, Biochem. Pharmacol., 2001, 61, 373–379 Search PubMed.
- K. Akira, M. Amano, F. Okajima, T. Hashimoto and S. Oikawa, Biol. Pharm. Bull., 2006, 29, 75–81 Search PubMed.
- P. Verbeke, G. E. Siboska, B. F. Clark and S. I. Rattan, Biochem. Biophys. Res. Commun., 2000, 276, 1265–1270 Search PubMed.
- Y. S. Jung, B. Y. Joe, S. J. Cho and Y. Konishi, Bioorg. Med. Chem. Lett., 2005, 15, 1125–1129 Search PubMed.
- S. M. Culbertson, G. D. Enright and K. U. Ingold, Org. Lett., 2003, 5, 2659–2662 Search PubMed.
- G. Munch, Y. Taneli, E. Schraven, U. Schindler, R. Schinzel, D. Palm and P. Riederer, Journal of Neural Transmission - Parkinson's Disease and Dementia Section, 1994, 8, 193–208 Search PubMed.
- V. Jakus, M. Hrnciarova, J. Carsky, B. Krahulec and N. Rietbrock, Life Sci., 1999, 65, 1991–1993 Search PubMed.
- Y. Keita, M. Michailova, W. Kratzer, G. Worner, W. Worner and N. Rietbrock, Int. J. Clin. Pharmacol. Ther. Toxicol., 1992, 30, 441–442 Search PubMed.
- A. Stevens, J. Am. Optom. Assoc., 1995, 66, 744–749 Search PubMed.
- Y. Al-Abed, T. Mitsuhashi, H. Li, J. A. Lawson, G. A. FitzGerald, H. Founds, T. Donnelly, A. Cerami, P. Ulrich and R. Bucala, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2385–2390 Search PubMed.
- B. H. Wolffenbuttel, C. M. Boulanger, F. R. Crijns, M. S. Huijberts, P. Poitevin, G. N. Swennen, S. Vasan, J. J. Egan, P. Ulrich, A. Cerami and B. I. Levy, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4630–4634 Search PubMed.
- D. L. Price, P. M. Rhett, S. R. Thorpe and J. W. Baynes, J. Biol. Chem., 2001, 276, 48967–48972 CrossRef CAS.
- G. L. Bakris, A. J. Bank, D. A. Kass, J. M. Neutel, R. A. Preston and S. Oparil, Am. J. Hypertens., 2004, 17, 23–30 Search PubMed.
- W. C. Little, M. R. Zile, D. W. Kitzman, W. G. Hundley, T. X. O'Brien and R. C. Degroof, J. Card. Failure, 2005, 11, 191–195 Search PubMed.
- D. Susic, J. Varagic, J. Ahn and E. D. Frohlich, Curr. Opin. Cardiol., 2004, 19, 336–340 Search PubMed.
- J. M. Forbes, L. T. Yee, V. Thallas, M. Lassila, R. Candido, K. A. Jandeleit-Dahm, M. C. Thomas, W. C. Burns, E. K. Deemer, S. R. Thorpe, M. E. Cooper and T. J. Allen, Diabetes, 2004, 53, 1813–1823 Search PubMed.
- R. Candido, J. M. Forbes, M. C. Thomas, V. Thallas, R. G. Dean, W. C. Burns, C. Tikellis, R. H. Ritchie, S. M. Twigg, M. E. Cooper and L. M. Burrell, Circ. Res., 2003, 92, 785–792 Search PubMed.
- S. Vasan, P. Foiles and H. Founds, Arch. Biochem. Biophys., 2003, 419, 89–96 Search PubMed.
- S. Vasan, X. Zhang, X. Zhang, A. Kapurniotu, J. Bernhagen, S. Teichberg, J. Basgen, D. Wagle, D. Shih, I. Terlecky, R. Bucala, A. Cerami, J. Egan and P. Ulrich, Nature, 1996, 382, 275–278 Search PubMed.
- G. B. Sajithlal, P. Chithra and G. Chandrakasan, Biochem. Pharmacol., 1998, 56, 1607–1614 CrossRef CAS.
- J. L. Wilkinson-Berka, D. J. Kelly, S. M. Koerner, K. Jaworski, B. Davis, V. Thallas and M. E. Cooper, Diabetes, 2002, 51, 3283–3289 Search PubMed.
- J. M. Forbes, T. Soulis, V. Thallas, S. Panagiotopoulos, D. M. Long, S. Vasan, D. Wagle, G. Jerums and M. E. Cooper, Diabetologia, 2001, 44, 108–114 Search PubMed.
- H. Y. Kim and K. Kim, J. Agric. Food Chem., 2003, 51, 1586–1591 CrossRef CAS.
- M. S. Ahmad and N. Ahmed, J. Nutr., 2006, 136, 796–799.
- M. Yoshikawa, Y. Pongpiriyadacha, A. Kishi, T. Kageura, T. Wang, T. Morikawa and H. Matsuda, Yakugaku Zasshi, 2003, 123, 871–880 CrossRef CAS.
- H. Y. Kim, B. H. Moon, H. J. Lee and D. H. Choi, J. Ethnopharmacol., 2004, 93, 227–230 CrossRef CAS.
- C. L. Hsieh, Y. C. Lin, W. S. Ko, C. H. Peng, C. N. Huang and R. Y. Peng, J. Ethnopharmacol., 2005, 102, 357–363 Search PubMed.
- D. S. Jang, J. M. Kim, Y. M. Lee, Y. S. Kim, J. H. Kim and J. S. Kim, Chem. Pharm. Bull., 2006, 54, 1315–1317 CrossRef CAS.
- J. M. Kim, Y. M. Lee, G. Y. Lee, D. S. Jang, K. H. Bae and J. S. Kim, Arch. Pharmacal Res., 2006, 29, 821–825 Search PubMed.
- K. S. Kang, H. Y. Kim, N. Yamabe, R. Nagai and T. Yokozawa, Biol. Pharm. Bull., 2006, 29, 1678–1684 Search PubMed.
- R. Suzuki, Y. Okada and T. Okuyama, J. Nat. Prod., 2003, 66, 564–565 CrossRef CAS.
- R. Suzuki, Y. Okada and T. Okuyama, Chem. Pharm. Bull., 2003, 51, 1186–1188 Search PubMed.
- R. Suzuki, M. Iijima, Y. Okada and T. Okuyama, Chem. Pharm. Bull., 2007, 55, 153–155 CrossRef CAS.
- L. Wirasathien, T. Pengsuparp, R. Suttisri, H. Ueda, M. Moriyasu and K. Kawanishi, Phytomedicine, 2006 Search PubMed.
- T. Nakagawa, T. Yokozawa, Y. A. Kim, K. S. Kang and T. Tanaka, Am. J. Chin. Med., 2005, 33, 817–829 Search PubMed.
- T. Yokozawa, A. Satoh, T. Nakagawa and N. Yamabe, Am. J. Chin. Med., 2006, 34, 307–321 Search PubMed.
- H. A. Jung, N. Y. Yoon, S. S. Kang, Y. S. Kim and J. S. Choi, J. Pharm. Pharmacol., 2008, 60, 1227–1236 Search PubMed.
- E. H. Lee, D. G. Song, J. Y. Lee, C. H. Pan, B. H. Um and S. H. Jung, Biol. Pharm. Bull., 2008, 31, 1626–1630 Search PubMed.
- T. Yokozawa, H. Y. Kim, E. J. Cho, N. Yamabi and J. S. Choi, J. Nutr. Sci. Vitaminol (Tokyo), 2003, 49, 87–93 Search PubMed.
- T. Kiho, S. Usui, K. Hirano, K. Aizawa and T. Inakuma, Biosci., Biotechnol., Biochem., 2004, 68, 200–205 CrossRef CAS.
- T. Hanamura, T. Hagiwara and H. Kawagishi, Biosci., Biotechnol., Biochem., 2005, 69, 280–286 Search PubMed.
- S. Rout and R. Banerjee, Bioresour. Technol., 2007, 98, 3159–3163 Search PubMed.
- K. Rutter, D. R. Sell, N. Fraser, M. Obrenovich, M. Zito, P. Starke-Reed and V. M. Monnier, Int. J. Vitam. Nutr. Res., 2003, 73, 453–460 CrossRef.
- T. Nakagawa, T. Yokozawa, K. Terasawa, S. Shu and L. R. Juneja, J. Agric. Food Chem., 2002, 50, 2418–2422 CrossRef CAS.
- N. Lunceford and A. Gugliucci, Fitoterapia, 2005, 76, 419–427 CrossRef.
- T. Yokozawa and T. Nakagawa, Food Chem. Toxicol., 2004, 42, 975–981 CrossRef CAS.
- P. Hegde, G. Chandrakasan and T. Chandra, J. Nutr. Biochem., 2002, 13, 517 Search PubMed.
- Y. Morimitsu, K. Yoshida, S. Esaki and A. Hirota, Biosci., Biotechnol., Biochem., 1995, 59, 2018–2021 Search PubMed.
- H. Lou, H. Yuan, Y. Yamazaki, T. Sasaki and S. Oka, Planta Med., 2001, 67, 345–349 CrossRef CAS.
- Y. Okada, A. Ishimaru, R. Suzuki and T. Okuyama, J. Nat. Prod., 2004, 67, 103–105 CrossRef CAS.
- T. Nagasawa, N. Tabata, Y. Ito, N. Nishizawa, Y. Aiba and D. D. Kitts, Mol. Cell. Biochem., 2003, 249, 3–10 Search PubMed.
- D. Cervantes-Laurean, D. D. Schramm, E. L. Jacobson, I. Halaweish, G. G. Bruckner and G. A. Boissonneault, J. Nutr. Biochem., 2006, 17, 531–540 Search PubMed.
- I. Bousova, J. Martin, L. Jahodar, J. Dusek, V. Palicka and J. Drsata, J. Pharm. Biomed. Anal., 2005, 37, 957–962 CrossRef CAS.
- C. H. Wu and G. C. Yen, J. Agric. Food Chem., 2005, 53, 3167–3173 CrossRef CAS.
- H. Matsuda, T. Wang, H. Managi and M. Yoshikawa, Bioorg. Med. Chem., 2003, 11, 5317–5323 CrossRef CAS.
- B. Manuel y Keenoy, J. Vertommen and I. De Leeuw, Diabetes Nutr. Metab., 1999, 12, 256–263 Search PubMed.
- C. Y. Lo, S. Li, D. Tan, M. H. Pan, S. Sang and C. T. Ho, Mol. Nutr. Food Res., 2006, 50, 1118–1128 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |