Multistage carcinogenesis process as molecular targets in cancer chemoprevention by epicatechin-3-gallate
Min-Hsiung
Pan
*a,
Yi-Siou
Chiou
ab,
Yin-Jen
Wang
b,
Chi-Tang
Ho
c and
Jen-Kun
Lin
*d
aDepartment of Seafood Science, National Kaohsiung Marine University, No.142, Haijhuan Rd., Nanzih District, Kaohsiung, 81143, Taiwan. E-mail: mhpan@mail.nkmu.edu.tw; Fax: (+886)-7-361-1261; Tel: (+886)-7-361-7141 ext. 3623
bDepartment of Environmental and Occupational Health, National Cheng Kung University Medical College, Tainan, 704, Taiwan
cDepartment of Food Science, Rutgers University, New Brunswick, New Jersey 08901-8520, USA
dGraduate Institute of Biochemistry and Molecular Biology, College of Medicine, National Taiwan University, No. 1, Section 1, Jen-ai Road, Taipei, Taiwan. E-mail: jklin@ha.mc.ntu.edu.tw; Fax: (+886)-2-2391-8944; Tel: (+886)-2-2356-2213
First published on 18th January 2011
Abstract
The consumption of green tea has long been associated with a reduced risk of cancer development. (−)-Epicatechin-3-gallate (ECG) or (−)-epigallocatechin-3-gallate (EGCG) are the major antioxidative polyphenolic compounds of green tea. They have been shown to exert growth-inhibitory potential of various cancer cells in culture and antitumor activity in vivo models. ECG or EGCG could interact with various molecules like proteins, transcription factors, and enzymes, which block multiple stages of carcinogenesis via regulating intracellular signaling transduction pathways. Moreover, ECG and EGCG possess pharmacological and physiological properties including induction of phase II enzymes, mediation of anti-inflammation response, regulation of cell proliferation and apoptosis effects and prevention of tumor angiogenesis, invasion and metastasis. Numerous review articles have been focused on EGCG, however none have been focused on ECG despite many studies supporting the cancer preventive potential of ECG. To develop ECG as an anticarcinogenic agent, more clear understanding of the cell signaling pathways and the molecular targets responsible for chemopreventive and chemotherapeutic effects are needed. This review summarizes recent research on the ECG-induced cellular signal transduction events which implicate in cancer management.
1. Introduction
Tea beverages are brewed from the Camellia sinensis plant (leaves) and have been consumed in China for nearly 5000 years.1 Of the total amount of tea undergone different manufacturing processes produced and consumed globally, 78% is black tea, 20% is green tea, and <2% is oolong tea.2 Black tea is consumed primarily in Western and some Asian countries, whereas green tea is consumed primarily in China, Japan, India, and a few countries in North Africa and the Middle East. The production and consumption of oolong tea and pu-erh tea are confined to southeastern China and Taiwan.3 In recent years, many studies from our and other laboratories have shown that green tea, oolong tea, black tea and pu-erh tea contains several tea catechin components; the major catechins in tea are (−)-epicatechin (EC), (−)-epicatechin-3-gallate (ECG), (−)-epigallocatechin (EGC), and (−)-epigallocatechin-3-gallate (EGCG).4–7 It is generally agreed that much of the cancer chemopreventive effects of green tea are due to its catechin compounds. Besides tea leaves, litchi (Litchi chinensis Sonn.) peel also contains a large amount of catechins, such as EGC, EC and ECG.8,9 Several studies indicated that litchi had good antioxidant10 and anticancer activities.11,12 Although these studies did not give a direct link between the activities of ECG and litchi peel, however, there was a strong possibility that ECG might be a major contributor to the cancer preventive efficacy in litchi peel.Vaidyanathan et al.13 have reported that ECG is absorbed by monocarboxylate transporter (MCT) resulting in accumulation of ECG in the lumen (75–300 μM) and this accumulated concentration is higher than EGCG or other catechins. It is known that only 0.1% of EGCG is bioavailable (absorbed) after an intragastric administration of green tea;14 on the other hand, ECG has higher hydrophobicity and preferentially accumulates in various cells compared with EGCG. Additionally, compared to EGCG, ECG is less cytotoxic, and relatively stable at the intracellular level.15 Thus, ECG may be more potent than EGCG in bioavailability and anti-cancer potential. Both EGCG and ECG are the most concentrated catechins in green tea, and are believed to be responsible for the anti-inflammatory/antioxidant activities of green tea.16 Green tea catechins have anticancer potential with multi-targets and multi-functions, and are non-toxic.17 Ravindranath et al. showed that the ECG inhibited the growth of human tumor cells with an equal or more potency than EGCG in gender-based carcinomas.18,19 On the other hand, ECG has been shown to be more susceptible to degradation than EGCG during storage of tea leaves20 and studies showed that ECG was not detected or detected at a very low level in human plasma and urine samples,21 suggesting that ECG could be conjugated with glucuronic acid and/or sulfate in plasma22,23 in animal models.24 Many literature and reviews display the cancer preventive effects of EGCG; only a few studies discussed the usefulness of ECG. Therefore, in this review, we focus on the potential anticarcinogenic and chemopreventive abilities of ECG, and discuss mechanisms on the modulation of cellular signaling events by ECG.
2. Chemopreventive potential of ECG in multi-stage carcinogenesis
Cancer is generally considered as uncontrolled cell division that results in the aggregation of cells to form tumors. There are many factors which are involved in the pathogenesis of cancer, such as: (1) individuals that engaged in risk-taking behaviors or lifestyles (e.g. smoking, use of snuff, and lack of proper diet like high in meat and low in fruits and vegetables);25 (2) exposure to known carcinogens (e.g. heavy metals of chromium);26 and (3) genetic mutations to the development of cancer (e.g. familial adenomatous polyposis,27etc.) On this basis, cancer results from a multistage carcinogenesis process in which distinct molecular and cellular alterations that involves three stages: initiation (normal cell → transformed or initiated cell), promotion (initiated cell → preneoplastic cell), and progression (preneoplastic cell → neoplastic cell).Initiation is a result of rather rapid and irreparable process to the cell, which includes the uptake of a carcinogenic agent and its distribution and transport to organs and tissues by its metabolic activation and the subsequent covalent interaction with target cell DNA, leading to stable genotoxic damage. The transformed cells undergo many changes to form preneoplastic cells. In contrast to initiation, tumor promotion process is not rapid, and oxidative stress and chronic inflammatory are key components in promoting tumor proliferation and angiogenesis which is necessary for solid tumor growth.28 Progression involves the gradual conversion of tumor cells to the invasive cells, leading to increase metastatic potential. Each of these progression processes (angiogenesis→ invasion→ metastasis) involves rate-limiting steps that are influenced by non-malignant cells of the tumour microenvironment.29
Tumour microenvironment involved many factors such as the reactive oxygen and nitrogen species (ROS and RNS), hypoxia, cytokines, growth factors, vascular endothelial growth factor (VEGF) and matrix metalloproteinase (MMP). The microenvironmental factors were produced by cancer cells, endothelial cells, stoma fibroblasts and a variety of bone marrow-derived cells (BMDCs).29,30 It can be suggested that three major types of chemopreventive agents are: (1) inhibitors for the formation of various carcinogens; (2) blocking agents to activate detoxification, to induce antioxidant enzymes, to reduce inflammatory responses and to decrease tumor cell growth by inducing apoptosis and/or cell cycle arrest; (3) suppressing agents to restrain the tumor cells from promotion and progression by destroying one or more cell signaling pathways. Therefore, chemopreventive agent in addition to known mechanism of action, it should have the following characteristics of little or no toxic effects; high efficiency; capability of oral administration; and low cost.31 Overall, ECG is an ideal chemopreventive agent (Fig. 1) which is known to reverse various intracellular signal transduction pathways by blocking or modulating the molecular expression during carcinogenesis processes (Fig. 2).
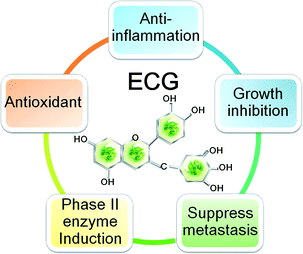 | ||
| Fig. 1 Chemoprevention and chemotherapy effect of ECG. | ||
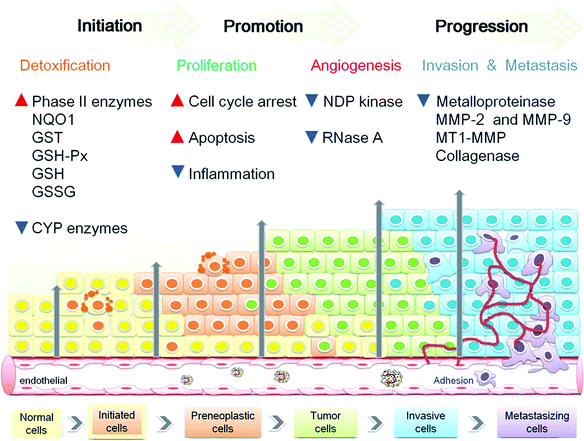 | ||
| Fig. 2 Schematic represents of chemopreventive targets and efficiency of ECG on multiple stage carcinogenesis. | ||
3. ECG block initiation stage in carcinogenesis process
Inhibition of metabolic and induction of phase II detoxifying/oxidant enzymes
Chemicals from dietary and environmental sources undergo oxidative metabolism by phase I enzymes, a major part of the cytochrome P450 monooxygenases superfamily, to get converted to polar (water-soluble) metabolites, which are subsequently excreted viaconjugation reactions catalyzed by phase II detoxifying enzymes, such as glutathione-S-transferase (GST), glutathione peroxidase (GSH-Px), superoxide dismutase (SOD), heme oxygenase-1 (HO-1) and NAD(P)H:quinone oxidoreductase 1 (NQO1), thus preventing carcinogens attacking cellular DNA and initiating tumorigenesis.32 In the absence of phases II enzymes, the metabolically active carcinogens can form a covalent adduct with DNA, resulting irreversible genotoxic damages. Irreparable damage leads to mutations in critical genes involved in growth, proliferation, and apoptosis, resulting in initiation and subsequent development of cancer. Several studies have demonstrated that ECG inhibited CYP450 isoforms implicated in rat CYP1A1/2,33 and human CYP3A4, CYP2A6, CYP2C19 and CYP2E134 in PB- or 3-methylcholanthrene-treated and B[a]P, PhIP and AFB1-treated CYP enzymes. Besides blocking the activation of CYP enzymes, ECG was shown to abrogate up to 50% benzo[a]pyrene (B[a]P)-diol epoxide-DNA adduct formation in B[a]P-treated BEAS-2B cells; inhibiting ODC activity and ∼20–30% free radical generation in 12-O-tetradecanoylphorbol-13-acetate (TPA)-stimulated 2C5 cells and HL-60 cells, respectively; increasing GST and NQO1 activity which could contribute to the inhibition of B[a]P-induced cell transformation ability in the anchorage-independent growth assay.35 In hydrogen peroxide (H2O2)-stimulated human bladder urothelial cells (TCCSUP and T24 BlCa), ECG could protect against oxidative stress/damage and cell death by reducing ROS production.36 Moreover, ECG also inhibited H2O2 production and ERK phosphorylation that preventing UVA-induced damage in keratinocytes (HaCaT cells).37 Besides, Chen et al. demonstrated that co-treatment with ECG and EGCG showed a synergistically protective effect compared to nongalloylated catechins by significantly increased cell viability, decreased lipid peroxidation and protected cell membrane against damage in lead-exposed HepG2 cells.38 These data indicated that ECG would potentiate antioxidant and anti-ageing capability. Protecting cells from oxidative damage not only can directly scavenge reactive oxygen species (ROS) but also enhance the body's antioxidant enzymes by inducing de novo expression of genes that encode detoxifying/antioxidant enzymes. Recent studies have demonstrated that ECG suppressed cellular lipid peroxidation through decreased thiobarbituric acid reactive substances (TBARS) accumulation, glutathione peroxidase (GSH-Px) activity and GSSG content, and increased GSH level, as well as reduced cytotoxicity in tert-butylated hydroperoxide (t-BOOH)-exposed HepG2 cells.39 It has been shown that the reduced oxidative stress by activation of nuclear factor erythroid 2 p45 (NF-E2)-related factor 2 (Nrf2) signaling could regulate antioxidant enzyme expression.40 Therefore, Nrf2 signaling is considered as an important molecular target for cancer prevention.41 Although, ECG can increase phase II enzymes, it is not clear whether this effect is due to the modulation of Nrf2 signaling.4. ECG block promotion stage in carcinogenesis process
Anti-inflammation efficacy
Chronic inflammation and infection are causally linked to multistage carcinogenic process.42 The inflammation processes lead to the up-regulation of a series of enzymes and signaling proteins in infected tissues and cells. These proinflammatory enzymes include the inducible forms of nitric oxide synthase (iNOS) and cyclooxygenase (COX-2), responsible for elevated levels of nitric oxide (NO) and prostaglandins (PGE2), respectively, and pro-inflammatory cytokines such as interleukin-1 (IL-1), IL-6 and tumor necrosis factor-α (TNF-α), have been known to be involved directly or indirectly in carcinogenesis, especially in the promotion and progression stages.43,44 In addition, more and more evidence suggests the role for chemokines in cancer development, such as the expression of adhesion molecules, the secretion of proteinases, inhibition of apoptosis, and angiogenesis.45 Thus, blocking inflammation signaling is usually recognized as potential mode for chemoprevention. Hong et al. have reported that ECG suppresses COX-2 and LOX activity, PGE2 production, and TBX and HHT formation in human colonic mucosa and tumor tissues.46ECG revealed anti-inflammatory effects by decreasing H2O2-induced intracellular ROS generation and impeding the phosphorylation of EGFR and ERK1, and suppressing MUC5AC overexpression.47ECG inhibited phosphorylation of p38 MAPK and ERK, and attenuated IL-17 receptor expression which results in preventing IL-17A-mediated CCL20 production.48 Besides inhibiting 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced inflammation (edema) in mouse ears,49ECG was also shown to significantly reverse interleukin-6 (IL-6) and to reduce transthyretin (TTR) and retinol binding protein (RBP) synthesis and secretion that could also be dependent on their antioxidant activities in human hepatoma cell line HepG2.50IL-6 is believed to modulate the acute-phase protein synthesis and to down-regulate the negative acute-phase protein levels (TTR and RBP) in various inflammatory states.51,52 Studies have shown that antioxidants may attenuate the acute-phase response. The acute-phase response is a collective term referring to the constellation of host defense mechanisms induced in response to inflammation and infection.53 Therefore, the inhibitory activity of ECG toward inflammation may include the activation of Nrf2 signaling pathways.Cell cycle regulation and growth inhibition
The progression of the cell cycle is regulated by cyclin-dependent kinase (CDK) molecules and cyclins, Cdk inhibitors (CDKIs), and check point kinases (Chk 1 and Chk 2), which drive the cell from one phase to the next and maintenance of cell growth and differentiation.54 Uncontrolled cell proliferation is characteristic of transformed and malignant cells. Therefore, the modulation of cell cycle progression is one of the important strategies for chemoprevention in the multistage carcinogenesis. Most of earlier literature indicates that ECG treatment inhibited cell growth in various transformed cancer cell lines including prostate, pancreas, colon, liver, lung, breast, melanoma and tongue in a dose- and time-dependent manner.18,55–61ECG has been reported to suppress abnormal cell growth and survival of lung tissues from B[a]P-induced lung carcinogenesis by blocking growth factor or inducing apoptosis-associated signal transduction molecular such as p53 and its downstream target genes such as Bcl-2, Bax, CDKI (p21 and p27), along with H-ras, c-myc, cyclinD1, at different time points.62 The anti-proliferation ability of ECG comes from arrest in G1 phase of the cell cycle progression via down-regulation of the β-catenin/T-cell factor (TCF)-mediated cyclin D1 gene transcription in SCC7cells.63ECG has been reported to inhibit proliferation and growth of cancerous cells by down-regulating NUDT6 and hnRNP B1 mRNA expression in HCT11664 and A549 cells,65 respectively. Additionally, ECG may also inhibit 5 α-reductase66 and fatty acid synthase (FAS)67,68 activity that are important enzymes involved in cellular metabolism and growth regulation. The anti-proliferation effect not only modulates by proliferative gene, but also regulates through growth factor stimulation.69 Most growth factors bind to specific receptors, like PDGF (platelet derived growth factor), fibroblast growth factor-2 (FGF-2) and epidermal growth factor (EGF), that can act, besides stimulation of many types of cells to divide, also stimulate cell growth, survival, differentiation, or migration, depending on the environment and the cell type.70,71Studies indicate that estrogen receptor (ER) cross-talks with growth factor resulting in induced tumor growth and survival via mediating intracellular kinase cascades and activating downstream transcription factors, which act coordinately to regulated target genes expression involved in multiple carcinogenesis processes.72–74ECG has been shown to inhibit ER-dependent breast cancer cell proliferation (MCF-7) and retarded tumor growth in immature female C57BL/6 mice by blocking ER activity.75 Moreover, ECG may also inhibit the PDGF-BB-induced tyrosine phosphorylation of PDGF β receptor (PDGF-Rβ) and colony formation on the anchorage-independent growth of A172 glioblastoma cells.76 Recent reports indicated that activator protein 1 (AP-1) transcriptional activity can be stimulated by various signaling pathways that include external signals (i.e., growth factors and tyrosine kinase receptors) and cellular downstream kinase pathways such as mitogen-activated protein kinases (MAPKs) of the extracellular-signal-regulated kinase (ERK), p38 and JUN amino-terminal kinase (JNK) families.77 AP-1 transcription factor regulates variety of genes that play a critical role in various cellular functions including inflammation, proliferation, transformation, survival and cell death; AP-1 activity has been associated with tumorigenesis of various types of cancers.78 Recently, ECG was shown to decrease AP-1 activity and cell proliferation in ras-transformed 30.7b cells.79 Furthermore, recent data suggested that ECG might exert anti-proliferation effects by inhibiting P-glycoprotein (P-gp) efflux pump activity and enhancing intracellular accumulation of P-gp substrates (e.g.daunorubicin and rhodamine-123) in the multidrug-resistant cell lines CH(R) C5,80 BEL-7404/DOX81 and KB-C2,82 and thereby increased anticancer efficiency.
Apoptosis mediation
Apoptosis (programmed cell death) is a multi-step, multi-pathway and highly ordered process that is a necessary part of the development and homeostasis of multicellular organisms. Apoptosis pathways can be initiated by a variety of stimuli, including genotoxic compounds and various environmental stresses (e.g. oxidative stress and irradiation) at the plasma membrane by death receptor ligation (extrinsic pathway) or at the mitochondria (intrinsic pathway). In both pathways, induction of apoptosis leads to activation of initiator caspases and then activate executioner caspases, which cleave the death substrates (like poly(ADP-ribose) polymerase (PARP)) and eventually result in DNA fragmentation.83,84Induction of apoptosis serves as a protective mechanism against the development of diseases such as cancer by eliminating genetically damaged cells or unwanted cells that have improperly been induced to divide.85 Cancerous cells escape from apoptosis through the overexpression of growth-promoting oncogenes and anti-apoptotic proteins such as Ras and Bcl-2 or by down-regulation or mutation of pro-apoptotic proteins such as p53 that promote neoplastic cell proliferation and tumorigenesis.86 Therefore, the activation of pro-apoptotic protein in cancer cells is one of the important strategies for cancer prevention. ECG has been reported to induce apoptosis in NCI-H460 cells by increasing the expression of p53 and reducing the expression of Bcl-2, but protein levels of H-ras and c-Myc remained unchanged.87Several reports revealed that non-steroidal anti-inflammatory drug (NSAID) activated gene (NAG-1) is a pro-apoptotic and antitumorigenic gene,88 which regulated by several transcription factors such as p53,89,90 activating transcription factor 3 (ATF3)91 and early growth response gene-1 (EGR-1).92 In HCT116 cells, ECG enhanced NAG-1 expression via mediated ATF3 transcriptional activity and induced TSP1 expression resulting in PARP cleavage and apoptosis, suggesting that ECG acted by a p53-independent pathway.93 Previous reports also indicated that EGR-1 acted as a tumor suppressor gene by directly binding to p53,94NAG-195 and phosphatase and tensin homolog (PTEN)96 promoters. Interestingly, ATF3 promoter activity is regulated by a variety of transcription factors, including NF-κB,97 EGR-198 and ATF/CRE.99 Similarly, ECG may also induce ATF3 and NAG-1 expression by increased EGR-1 transcriptional capability, these results displayed that ROS participated in the ECG-induced ATF3 expression but not ECG-induced NAG-1 expression.100 Simon et al. have shown that intracellular accumulation of ROS induced apoptosis by stimulating cytochrome c release to the cytosol that triggered caspase activation in many biological systems.101 Accordingly, ROS and mitochondria play an important role in proapoptotic activity. Previously studies showed that ECG could increased ROS formation and mitochondrial depolarization, thereby inducing apoptosis in DU145 cells.102 Moreover, ECG also induced caspase-3 activity, DNA fragmentation, H2O2 generation and apoptosis in the carcinoma HSC-2 cells.103 Therefore, recent studies suggested that ECG contributed to pro-oxidative activity which played an important role in proapoptotic pathways. In another study, ECG effectively suppressed the growth and survival of KATO III cells by blocking okadaic acid-induced tumor necrosis factor-α (TNF-α) release and initiating apoptosis by inducing DNA fragmentation.104 Accumulating studies exhibited that activation of TNF-α signaling pathway might lead to cell proliferation and anti-apoptosis through activated downstreamMAPK kinase and transcription factors such as NF-κB and AP-1.105 Although ECG may induce apoptosis by triggering various molecules, this beneficial efficacy is still deficient in solid molecular mechanism and requires further investigation.
5. ECG blocks progression stage in carcinogenesis process
Anti-metastasis ability
Cancer cells migrate from the primary tumor to distant organs and proliferate at the metastatic sites, are not only promoting cancer progression but also the major cause of death for cancer patients.106 At each metastatic step, the tumor cells can be modulated by microenvironmental factors, including interactions with variety of proteolytic enzymes (e.g. matrix metalloproteinases), growth factors (e.g.EGF), and cell-cell and cell-substrate adhesion molecules (e.g. ECM protein).29 Therefore, prevention of cancer metastasis is considered novel targets for therapy in common malignancies. Collagenases (e.g. MMP-1, MMP-13) and gelatinases (e.g. MMP-2, MMP-9) are known as matrix metalloproteinases (MMPs), which act as important mediators of ECM degradation involved in controlling tumor invasion.107,108ECG has been reported to inhibit HT1080 cells invasion by blocking matrix metalloproteinases activity-9 (MMP-9) and -2 (MMP-2) activities in the absence of cytotoxicity.109 Likewise, ECG also reduced thrombin-induced activation of MMP-2 by direct inhibiting membrane-type matrix metalloproteinases-1 (MT1-MMP) activity, which led to the inhibition of invasion of vascular smooth muscle cells (VSMCs).110 Moreover, ECG has stronger inhibitory effect than EGCG on both eukaryotic and prokaryotic cell-derived collagenase111 and MMP-7 (Matrilysin)112 activities.Numerous laboratories have demonstrated that hepatocyte growth factor (HGF) plays a crucial role in cancer cell migration, matrix adhesion, invasion, proliferation and angiogenesis, via the phosphorylation of the c-met tyrosine kinase and activation of downstream signaling molecules including AKT, Ras/MAPK and the JAK/STAT pathway.113–115ECG has been shown to inhibit HGF-induced cell motility and scattering by blocking HGF-stimulated Met, ERK and AKT phosphorylation.116,117 According to a recent reports, EGCG may be more effective than ECG in preventing tumor cell angiogenesis through abolish nucleoside diphosphate kinase (NDPK-B)118 and ribonuclease A (RNase A)119 activities, which are associated with tumor-induced metastatic process.120,121 Additionally, ECG could impair the adhering and/or spreading of mouse lung carcinoma 3LL and melanoma B16F10 cells to the fibronetin and endothelial cells,122,123 which play a critical role in the process of metastatic tumor dissemination.124 Taken together, these findings suggest that ECG possesses anti-metastasis property by regulating multiple intracellular signaling molecules.
6. Discussion
Table 1 summarizes various molecular targets of ECG as a cancer chemopreventive agent. The efficacy of ECG is also compared to EGCG in various systems. Several studies have demonstrated that EGCG and ECG reduced the carcinogenic formation by interacting with microsomal cytochrome P450 enzyme proteins and impairing electron transfer. The inhibition of carbon monoxide-reduced cytochrome P450 binding by EGCG was higher than ECG. Furthermore, EGCG and ECG were potent inducer of phase II enzymes. Activation of these enzymes was accompanied by significant attenuation of lipid peroxidation and against oxidative stress.| Molecular targets | Experimental models | Reference | ¦ Outcomes | |
|---|---|---|---|---|
| Studied type | Dose | |||
| CYP enzymes | ||||
| ↓CYP45 activity | PB- or 3-methylcholanthrene-treated rats | 0.2–100 mM | 33 | EGCG > ECG |
| ↓CYP1A1/2, CYP3A4, CYP2A6, CYP2C19, and CYP2E1 activity | B[a]P, PhIP and AFB1 treated human CYP enzymes | IC50 = 20, 35, 30 μM | 34 | EGCG > ECG |
| Phase II detoxification and antioxidant enzymes | ||||
| ↓B[a]P DNA adduct formation;↓ODC activity; ↑NADPH:QR (NQO1);↑GST activity;↓Free radical | BEAS-2B cells; 2C5 cells; HL-60 cells, A427, RTE | 0.0001–1 mM | 35 | EGCG = ECG |
| ↓ROS (oxidative stress/damage) | H2O2-treated human bladder urothelial cells | 10–40 μg ml−1 | 36 | ECG > EGCG |
| ↓Hydrogen peroxide (H2O2) production; ↓ERK1/2 phosphorylation | UVA-induced HaCaT keratinocyte | 1–100 μM | 37 | ECG > EGCG |
| ↓GSH-Px activity;↓GSSG and TBARS level;↑GSH content | t-BOOH-treated HepG2 cells | 10–25 μM | 39 | EGCG > ECG |
| Pro-inflammatory mediators | ||||
| ↓COX and LOX activity;↓PGE2 production; ↓TBX and HHT formation | Colon mucosa and tumor from human | 30 μg ml−1 | 46 | ECG > EGCG |
| ↓ROS production;↓p-EGFR (tyr1068,1048,992 and 845); ↓Expression of protein and mRNA MUC5AC;↓p-ERK1/2 | H2O2–induced Passage-2 NHNE cells | 100 μM | 47 | ECG |
| ↑TTR and RBP levels | IL-6-stimulated HepG2 cells | 25 μM | 50 | EGCG > ECG |
| ↓Edema | TPA-treated mouse ear | 1 μmol | 49 | EGCG > ECG |
| ↓Chemokine CCL20 production;↓IL-17R expression ↓p38 and p-ERK | IL-17A-stimulated HGF cells | 50 μg ml−1 | 48 | EGCG > ECG |
| Growth inhibition and cell cycle arrest | ||||
| ↓NUDT6 expression | HCT-116 cells | 50 μM | 64 | EGCG > ECG |
| ↓hnRNP B1 mRNA level | A549 cells | IC50 = 50 μM | 65 | EGCG > ECG |
| ↓AP-1 activity | 30.7b Ras 12 cells | IC50 = 15 μM | 79 | EGCG > ECG |
| ↓H-ras, c-myc, cyclinD1 and Bcl-2 expression; ↑p21, p27, p53 and Bax expression | B[a]P-induced lung carcinogenesis | 4 μg | 62 | EGCG = ECG |
| ↓P-glycoprotein (P-gp) efflux pump activity | BEL-7404/DOX; CHRC5 cells; KB-C2 cells | 50–100 μM | 80–82 | EGCG > ECG |
| ↓CyclinD1 expression;↓β-catenin activity | SCC7 cells | 50 μM | 63 | ECG > EGCG |
| ↓PDGF-Rβ tyrosine kinase activity | A172 glioblastoma cells | 50 μM | 76 | ECG ≧ EGCG |
| ↓ERβ activity;↑Uterine peroxidase activity | MCF-7, C57BL/6 mice | 1 μM | 75 | ECG ≧ EGCG |
| ↓5 α-reductase activity | Rat liver and rat-1A cells | IC50 = 12 μM | 66 | EGCG > ECG |
| ↓Fatty acid synthase (FAS) activity | chicken liver FAS | IC50 = 42 μM | 67,68 | ECG > EGCG |
| Molecules of the apoptotic signaling pathway | ||||
| ↑p53 expression;↓Bcl-2 expression | NCI-H460 cells | 87 | ||
| ↑Caspase-3 activity;↑H2O2 production | HSC-2 cells | 125 μM | 103 | ECG > EGCG |
| ↑NAG-1 and ATF3 expression;↑Egr-1 activity | HCT-116 cells | 50 μM | 100 | EGCG = ECG |
| ↑Expression of NAG-1 and TSP-1 protein and mRNA; ↑ATF3 activity;↑cleavage of PARP | HCT-116 cells | 50 μM | 93 | ECG > EGCG |
| ↑ROS production;↑mitochondrial depolarization | DU145 cells | 100 μM | 102 | ECG > EGCG |
| ↓TNF-α release | KATO III cells | 26–500 μM | 104 | ECG > EGCG |
| Invasion and metastatic progression | ||||
| ↓MMP-2 and MMP9 activity | HT1080 cells | 100 μg ml−1 | 109 | ECG > EGCG |
| ↓MMP-2 and MT1-MMP activity | Thrombin-induced VSMCs | 30 μM | 110 | ECG > EGCG |
| ↓MMP-7 activity | IC50 = 0.47 μM | 132 | ECG > EGCG | |
| ↓↓phosphorylation of Met, ERK and Akt | MCF10A cells | 0.6∼5 μM | 116 | EGCG > ECG |
| ↓Phosphorylation of ERK and Akt | DU145 cells | 1∼10 μM | 117 | EGCG = ECG |
| ↓NDP kinase activity | MDA-MB-435 cells | −3.5 log M | 118 | EGCG > ECG |
| ↓Adhesion and/or spreading | 3LL or B16F10 cells | 122,123 | ||
| ↓Ribonuclease A (RNase A) enzymatic activity | Cu(II)-ECG complex | 4∼6.7 μM | 119 | EGCG > ECG |
| ↓Collagenase activity | Prokaryotic and eukaryotic cell | 100 μg ml−1 | 111 | ECG > EGCG |
However, ECG strongly protected the cell damage, cytotoxicity and chronic inflammatory, induced by H2O2 and UVA. Adachi et al.125 have reported that EGCG could suppress epidermal growth factor receptor (EGFR) activation by induce internalization of EGFRs into endosomes in colon cancer cells. In contrast, ECG may inhibit the dimerization or intracellular kinase phosphorylation and endocytosis of EGFR at the cell surface of NHNE cells, but does not change the number of cell surface-associated EGFR. Previous reports indicate that ECG has strong inhibitory effect on arachidonic acid metabolism in human colon mucosa and colon tumors, and prevention of inflammation-induced carcinogenesis. In inflammatory processes, EGCG treatment more effectively suppressed production of proinflammatory cytokines and phosphorylation of p38 and ERK compared to ECG treatment. In addition, EGCG can also potently enhance negative acute-phase protein (TTR and RBP) secretions and achieve anti-inflammation actions.
Several in vivo and in vitro studies revealed that EGCG and ECG could regulate growth inhibition and induce apoptosisvia modulation of different mechanisms. EGCG and ECG has been shown to reduce H-ras, c-myc, cyclinD1 and Bcl-2gene expression and increase p53 level in B[a]P-induced lung carcinogenesis, but no significant influence on H-ras and c-Myc expressions in a highly metastatic human lung cancer cell line NCI-H460. Moreover, some reports indicated that EGCG and ECG induced apoptosis was mediated via production of ROS (e.g.H2O2).
Although ECG has been shown to generate less H2O2 than EGCG, it is still found to have greater cytotoxicity to carcinoma HSC-2 cells than the normal HGF-2 fibroblasts. Comparisons with ECG, EGCG showed higher inhibition of cell growth and AP-1 activity due to the presence of a galloyl structure on the B ring. In other research, EGCG more markedly suppressed the proliferation and growth than ECGvia decreasing the proliferative gene NUDT6 level in HCT116 cells. However, ECG showed more effective in inducing apoptosis of DU145 and KATO III cells than EGCG by increasing ROS formation and TNF-α release, respectively. Besides, ECG seems to better inhibit cell growth by blocking β-catenin, PDGF-Rβ, ERβ and FAS activity in different experimental systems. In HCT116 cells, ECG showed stronger antitumorigenic activity than EGCG by activating transcription factors (e.g. Egr-1, ATF-3) mediated anti-cancer gene expression, including TSP-1 and NAG-1. Nevertheless, EGCG-induced NAG-1 expression is regulated by p53. Recently studies also indicate that ECG had the strongest anti-invasion activity by reducing MMP-2 and MMP-9 activity and their activation by a direct inhibition of MT1-MMP.
These studies suggest that ECG may be biologically more active than EGCG, and EGCG was not always the most potent chemopreventive agent among green tea catechins. While EGCG has been well studied and is known to have chemopreventive property in several cancer cells, but molecular mechanisms of ECG have not been well investigated. Therefore, research on the function of ECG is important for understanding its anti-tumor effect.
Previous studies found that ECG more effective than EGCG induced apoptosis and increased cell cycle arrest by inhibiting β-catenin signaling and cyclin D1 expression in SSC-7 cells.63 Interestingly, EGCG may affect anti-tumorigenic activity in a cyclin D1-independent manner.126EGCG induced apoptosis through increasing H2O2 generation, but not found in ECG-induced apoptosis in HSC-2 cells.103 It has been found that treatment with EGCG caused G1 arrest and apoptosis in LoVo cells, whereas ECG triggers just the former process.127 Another study displayed that ECG induced apoptosis of HCT116 cells by mediating NAG-1 expression viaATF3 in a p53-independent manner, but EGCG is involved in p53-induced NAG-1 expression.93 These are suggested by resent results, ECG seems to better modulate cell apoptosis in p53 mutant tumor cells. Therefore, these results suggest that ECG and EGCG display differences in anti-tumor mechanisms.
7. Conclusions
In green tea extract, the percentages of the main catechins are EGCG 10–15%, EGC 6–10%, and ECG 2–3%.128 However, high ECG (537.14 μg mL−1) occurs in some pu-erh tea129 and pu-erh green tea (EGCG 7.689% and ECG 9.890%, respectively).130 It is clear that ECG can interfere with multiple cell signaling pathways and has multiple targets within the cells, which are likely to interact together to reduce the risk of carcinogenesis (initiation, promotion and progression stages). These mechanisms include (a) inhibition of phase 1 CYP enzymes, (b) induction of phase II detoxification and antioxidant enzymes, (c) anti-inflammatory efficacy (d) arrest of cell cycle progression, (e) regulation of pro-apoptotic properties and (f) mediation of metastasis processes (Fig. 2 and 3). Many of the anti-carcinogenic affects of ECG may be due to its direct and/or indirect interaction with numerous molecular targets,131 such as NAG-1, AP-1, 5α-reductase and PDGF. Importantly, these growth inhibitions of ECG have been shown to sensitize cancer cells, but not in normal cells. Despite the regulation of intracellular signaling pathways, ECG may also inhibit RNase A and MMPs enzymatic activity via chelating copper and zinc metals, which are important cofactors for angiogenesis and metastasis. Furthermore, structure function analysis revealed that the gallate moiety of ECG is important for mediating these inhibitory effects which these acts may enhance chemoprevention ability.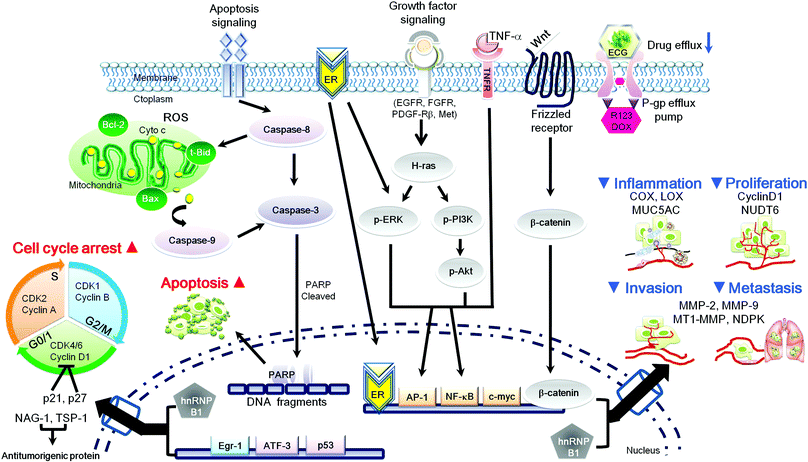 | ||
| Fig. 3 Schematic representation of ECG mediated intracellular signaling transduction pathways on carcinogenesis processes. ▲ Induction of signaling cascades by ECG-regulated; ▼ inhibition of signaling cascades by ECG-regulated. | ||
Abbreviations
| AP-1 | Activator protein-1 |
| ATF3 | Activating transcription factor 3 |
| BMDCs | Bone marrow-derived cells |
| CAMs | Cell adhesion molecules |
| CDK | Cyclin-dependent kinase |
| CDKIs | Cdk inhibitors |
| Chk | Check point kinases |
| COX-2 | Cyclooxygenase-2 |
| EC | (−)-Epicatechin |
| ECG | Epicatechin-3-gallate |
| EGCG | (−)-Epigallocatechin-3-gallate |
| EGC | (−)-Epigallocatechin |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| EGR-1 | Early growth response gene-1 |
| ER | Estrogen receptor |
| ERK | Extracellular-signal-regulated kinase |
| FAS | Fatty acid synthase |
| FGF-2 | Fibroblast growth factor-2 |
| GSH-Px | Glutathione peroxidase |
| GST | Glutathione-S-transferase |
| HGF | Hepatocyte growth factor |
| hnRNPB1 | Heterogeneous nuclear ribonucleoprotein B1 |
| HO-1 | Heme oxygenase-1 |
| H2O2 | Hydrogen peroxide |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| JNK | Jun amino-terminal kinase |
| MAPK | Mitogen activated protein kinase |
| MCT | Monocarboxylate transporter |
| MMP | Matrix metalloproteinase |
| MT1-MMP | Membrane-type matrix metalloproteinases-1 |
| NAG-1 | Non-steroidal anti-inflammatory drug (NSAID) activated gene-1 |
| NDPK-B | Nucleoside diphosphate kinase |
| NF-κB | Nuclear factor-κB |
| NO | Nitric oxide |
| NQO1 | NAD(P)H:quinone oxidoreductase 1 |
| Nrf2 | Nuclear factor erythroid 2 p45 (NF-E2)-related factor 2 |
| NUDT6 | Nudix (nucleoside diphosphate linked moiety X)-type motif 6 |
| PDGF | Platelet-derived growth factor |
| PI3K | Phosphatidylinositol-3-kinase |
| PGs | Prostaglandins |
| P-gp | P-Glycoprotein |
| PARP | Poly(ADP-ribose) polymerase |
| PTEN | Phosphatase and tensin homolog |
| RBP | Retinol binding protein |
| ROS | Reactive oxygen species |
| RNS | Reactive nitrogen species |
| RNase A | Ribonuclease A |
| SOD | Superoxide dismutase |
| TBARS | Thiobarbituric acid reactive substances |
| t-BOOH | tert-Butylated hydroperoxide |
| TCF | T-cell factor |
| TGF-β | Transforming growth factor- β |
| TIMP | Tissue inhibitor of metalloproteinase |
| TNF-α | Tumor necrosis factor- α |
| TPA | 12-O-Tetradecanoyl-phorbol-acetate |
| TTR | Transthyretin |
| VEGF | Vascular endothelial growth factor |
| VSMCs | Vascular smooth muscle cells |
References
- D. L. McKay and J. B. Blumberg, J. Am. Coll. Nutr., 2002, 21, 1 CAS.
- J. Ju, G. Lu, J. D. Lambert and C. S. Yang, Semin. Cancer Biol., 2007, 17, 395 CrossRef CAS.
- C. Han and Y. Gong, Wei Sheng Yan Jiu, 1999, 28, 343 Search PubMed.
- M. H. Ravindranath, V. Ramasamy, S. Moon, C. Ruiz and S. Muthugounder, Evid. Based. Complement Alternat. Med., 2009, 6, 523 Search PubMed.
- T. Ohe, K. Marutani and S. Nakase, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2001, 496, 75 CrossRef CAS.
- K. M. Ku, J. Kim, H. J. Park, K. H. Liu and C. H. Lee, J. Agric. Food Chem., 2010, 58, 345 CrossRef CAS.
- T. D. Way, H. Y. Lin, D. H. Kuo, S. J. Tsai, J. C. Shieh, J. C. Wu, M. R. Lee and J. K. Lin, J. Agric. Food Chem., 2009, 57, 5257 CrossRef CAS.
- T. Kuzuhara, M. Suganuma and H. Fujiki, Cancer Lett., 2008, 261, 12 CrossRef CAS.
- P. C. Q. J. M. G. Donglin Zhang, Postharvest Biol. Technol., 2000, 19, 165 CrossRef.
- X. Duan, G. Wu and Y. Jiang, Molecules, 2007, 12, 759 Search PubMed.
- X. Wang, Y. Wei, S. Yuan, G. Liu, Y. L. Zhang and W. Wang, Cancer Lett., 2006, 239, 144 CrossRef CAS.
- A. H. Xiong, W. J. Shen, L. Y. Xiao and J. H. Lv, Zhong Yao Cai, 2008, 31, 1533 Search PubMed.
- J. B. Vaidyanathan and T. Walle, J. Pharmacol. Exp. Ther., 2003, 307, 745 CrossRef CAS.
- L. Chen, M. J. Lee, H. Li and C. S. Yang, Drug Metab Dispos., 1997, 25, 1045 CAS.
- Y. Kawai, H. Tanaka, K. Murota, M. Naito and J. Terao, Biochem. Biophys. Res. Commun., 2008, 374, 527 CrossRef CAS.
- H. K. Na and Y. J. Surh, Food Chem. Toxicol., 2008, 46, 1271 CrossRef CAS.
- S. B. Moyers and N. B. Kumar, Nutr. Rev., 2004, 62, 204 Search PubMed.
- M. H. Ravindranath, T. S. Saravanan, C. C. Monteclaro, N. Presser, X. Ye, S. R. Selvan and S. Brosman, Evid. Based. Complement Alternat. Med., 2006, 3, 237 Search PubMed.
- M. H. Ravindranath, V. Ramasamy, S. Moon, C. Ruiz and S. Muthugounder, Evid. Based. Complement Alternat. Med., 2009, 6, 523 Search PubMed.
- M. Friedman, C. E. Levin, S. U. Lee and N. Kozukue, J. Food Sci., 2009, 74, H47 CrossRef CAS.
- M. J. Lee, Z. Y. Wang, H. Li, L. Chen, Y. Sun, S. Gobbo, D. A. Balentine and C. S. Yang, Cancer Epidemiol. Biomarkers Prev., 1995, 4, 393 CAS.
- M. J. Lee, P. Maliakal, L. Chen, X. Meng, F. Y. Bondoc, S. Prabhu, G. Lambert, S. Mohr and C. S. Yang, Cancer Epidemiol. Biomarkers Prev., 2002, 11, 1025 CAS.
- P. G. Pietta, P. Simonetti, C. Gardana, A. Brusamolino, P. Morazzoni and E. Bombardelli, BioFactors, 1998, 8, 111 Search PubMed.
- M. L. Mata-Bilbao, C. Andres-Lacueva, E. Roura, O. Jauregui, E. Escribano, C. Torre and R. M. Lamuela-Raventos, Br. J. Nutr., 2008, 100, 496 CAS.
- V. J. de, D. S. Jonker-Termont, E. M. van Lieshout, M. B. Katan and M. R. van der, Carcinogenesis, 2005, 26, 387.
- E. T. Snow, Pharmacol. Ther., 1992, 53, 31 CrossRef CAS.
- R. B. van der Luijt, C. M. Tops and H. F. Vasen, Ned. Tijdschr. Geneeskd., 2000, 144, 2007 Search PubMed.
- L. M. Coussens and Z. Werb, Nature, 2002, 420, 860 CrossRef CAS.
- J. A. Joyce and J. W. Pollard, Nat. Rev. Cancer, 2009, 9, 239 CrossRef CAS.
- J. A. Cook, D. Gius, D. A. Wink, M. C. Krishna, A. Russo and J. B. Mitchell, Semin. Radiat. Oncol., 2004, 14, 259 CrossRef.
- M. A. Morse and G. D. Stoner, Carcinogenesis, 1993, 14, 1737 CrossRef CAS.
- A. N. Kong, E. Owuor, R. Yu, V. Hebbar, C. Chen, R. Hu and S. Mandlekar, Drug Metab. Rev., 2001, 33, 255 CrossRef CAS.
- Z. Y. Wang, M. Das, D. R. Bickers and H. Mukhtar, Drug Metab Dispos., 1988, 16, 98 CAS.
- S. Muto, K. Fujita, Y. Yamazaki and T. Kamataki, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2001, 479, 197 CrossRef CAS.
- V. E. Steele, G. J. Kelloff, D. Balentine, C. W. Boone, R. Mehta, D. Bagheri, C. C. Sigman, S. Zhu and S. Sharma, Carcinogenesis, 2000, 21, 63 CrossRef CAS.
- C. H. Coyle, B. J. Philips, S. N. Morrisroe, M. B. Chancellor and N. Yoshimura, Life Sci., 2008, 83, 12 CrossRef CAS.
- C. C. Huang, J. Y. Fang, W. B. Wu, H. S. Chiang, Y. J. Wei and C. F. Hung, Arch. Dermatol. Res., 2005, 296, 473 CrossRef CAS.
- L. Chen, X. Yang, H. Jiao and B. Zhao, Toxicol. Sci., 2002, 69, 149 CrossRef CAS.
- C. Murakami, Y. Hirakawa, H. Inui, Y. Nakano and H. Yoshida, Biosci., Biotechnol., Biochem., 2002, 66, 1559 CrossRef CAS.
- A. K. Jaiswal, Free Radical Biol. Med., 2004, 36, 1199 CrossRef CAS.
- J. S. Lee and Y. J. Surh, Cancer Lett., 2005, 224, 171 CrossRef CAS.
- H. Ohshima and H. Bartsch, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1994, 305, 253 CrossRef CAS.
- C. Santangelo, R. Vari, B. Scazzocchio, B. R. Di, C. Filesi and R. Masella, Ann. Ist. Super. Sanita, 2007, 43, 394 Search PubMed.
- K. S. Zaenker, Contrib. Microbiol., 2006, 13, 232 Search PubMed.
- P. Proost, A. Wuyts and D. J. van, Int. J. Clin. Lab Res., 1996, 26, 211 CrossRef CAS.
- J. Hong, T. J. Smith, C. T. Ho, D. A. August and C. S. Yang, Biochem. Pharmacol., 2001, 62, 1175 CrossRef CAS.
- H. J. Kim, J. H. Ryu, C. H. Kim, J. W. Lim, U. Y. Moon, G. H. Lee, J. G. Lee, S. J. Baek and J. H. Yoon, Am. J. Respir. Cell Mol. Biol., 2009 Search PubMed.
- Y. Hosokawa, I. Hosokawa, K. Ozaki, T. Nakanishi, H. Nakae and T. Matsuo, Cell. Physiol. Biochem., 2009, 24, 391 CrossRef CAS.
- M. T. Huang, C. T. Ho, Z. Y. Wang, T. Ferraro, T. Finnegan-Olive, Y. R. Lou, J. M. Mitchell, J. D. Laskin, H. Newmark and C. S. Yang, Carcinogenesis, 1992, 13, 947 CrossRef CAS.
- M. A. el-Saadany, H. M. Rawel, J. Raila, M. S. el-Dashloty and F. J. Schweigert, Cell Biochem. Funct., 2008, 26, 95 CrossRef CAS.
- J. V. Castell, M. J. Gomez-Lechon, M. David, R. Fabra, R. Trullenque and P. C. Heinrich, Hepatology, 1990, 12, 1179 CrossRef CAS.
- P. C. Heinrich, J. V. Castell and T. Andus, Biochem. J., 1990, 265, 621 CAS.
- F. Ceciliani, A. Giordano and V. Spagnolo, Protein Pept. Lett., 2002, 9, 211 CrossRef CAS.
- I. Collins and M. D. Garrett, Curr. Opin. Pharmacol., 2005, 5, 366 CrossRef CAS.
- X. Tan, D. Hu, S. Li, Y. Han, Y. Zhang and D. Zhou, Cancer Lett., 2000, 158, 1 CrossRef CAS.
- L. Chen, X. Yang, H. Jiao and B. Zhao, Toxicol. Sci., 2002, 69, 149 CrossRef CAS.
- C. Chen, R. Yu, E. D. Owuor and A. N. Kong, Arch. Pharmacal Res., 2000, 23, 605 Search PubMed.
- T. M. Elattar and A. S. Virji, Anticancer Res., 2000, 20, 3459 CAS.
- G. Y. Yang, J. Liao, K. Kim, E. J. Yurkow and C. S. Yang, Carcinogenesis, 1998, 19, 611 CrossRef CAS.
- S. Okabe, M. Suganuma, M. Hayashi, E. Sueoka, A. Komori and H. Fujiki, Jpn. J. Cancer Res., 1997, 88, 639 CAS.
- S. Valcic, B. N. Timmermann, D. S. Alberts, G. A. Wachter, M. Krutzsch, J. Wymer and J. M. Guillen, Anti-Cancer Drugs, 1996, 7, 461 CAS.
- S. Manna, S. Mukherjee, A. Roy, S. Das and C. K. Panda, J. Nutr. Biochem., 2009, 20, 337 CrossRef CAS.
- Y. C. Lim, S. H. Lee, M. H. Song, K. Yamaguchi, J. H. Yoon, E. C. Choi and S. J. Baek, Eur. J. Cancer, 2006, 42, 3260 CrossRef CAS.
- M. Sukhthankar, C. K. Choi, A. English, J. S. Kim and S. J. Baek, J. Nutr. Biochem., 2010, 21, 98 CrossRef CAS.
- N. Fujimoto, N. Sueoka, E. Sueoka, S. Okabe, M. Suganuma, M. Harada and H. Fujiki, Int. J. Oncol., 2002, 20, 1233 CAS.
- S. Liao and R. A. Hiipakka, Biochem. Biophys. Res. Commun., 1995, 214, 833 CrossRef CAS.
- W. X. Tian, Curr. Med. Chem., 2006, 13, 967 CrossRef CAS.
- X. Wang, K. S. Song, Q. X. Guo and W. X. Tian, Biochem. Pharmacol., 2003, 66, 2039 CrossRef CAS.
- T. Kondo, S. Ezzat and S. L. Asa, Nat. Rev. Cancer, 2006, 6, 292 CrossRef CAS.
- R. D. Stanzel, S. Lourenssen, D. G. Nair and M. G. Blennerhassett, Am. J. Physiol Cell Physiol, 2010, 299, C805 CrossRef CAS.
- P. M. Comoglio, M. F. Di Renzo, G. Gaudino, C. Ponzetto and M. Prat, Am. Rev. Respir. Dis., 1990, 142, S16 Search PubMed.
- R. Schiff, S. A. Massarweh, J. Shou, L. Bharwani, S. K. Mohsin and C. K. Osborne, Clin. Cancer Res., 2004, 10, 331S CrossRef CAS.
- H. Lu, J. Li, D. Zhang, G. D. Stoner and C. Huang, Nutr. Cancer, 2006, 54, 69 Search PubMed.
- D. P. Edwards and V. Boonyaratanakornkit, Mol. Interventions, 2003, 3, 12 CrossRef CAS.
- M. G. Goodin, K. C. Fertuck, T. R. Zacharewski and R. J. Rosengren, Toxicol. Sci., 2002, 69, 354 CrossRef CAS.
- A. Sachinidis, C. Seul, S. Seewald, H. Ahn, Y. Ko and H. Vetter, FEBS Lett., 2000, 471, 51 CrossRef CAS.
- R. Eferl and E. F. Wagner, Nat. Rev. Cancer, 2003, 3, 859 CrossRef CAS.
- E. Shaulian, Cell. Signalling, 2010, 22, 894 CrossRef CAS.
- J. Y. Chung, C. Huang, X. Meng, Z. Dong and C. S. Yang, Cancer Res., 1999, 59, 4610 CAS.
- J. Jodoin, M. Demeule and R. Beliveau, Biochim. Biophys. Acta, Mol. Cell Res., 2002, 1542, 149 CrossRef CAS.
- G. Liang, A. Tang, X. Lin, L. Li, S. Zhang, Z. Huang, H. Tang and Q. Q. Li, Int. J. Oncol., 2010, 37, 111 CAS.
- S. Kitagawa, T. Nabekura and S. Kamiyama, J. Pharm. Pharmacol., 2004, 56, 1001 CrossRef CAS.
- M. O. Hengartner, Nature, 2000, 407, 770 CrossRef CAS.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495 CrossRef CAS.
- N. N. Danial and S. J. Korsmeyer, Cell, 2004, 116, 205 CrossRef CAS.
- G. Klein, Cell Death. Differ., 2004, 11, 13 CrossRef CAS.
- C. Ganguly, P. Saha, C. K. Panda and S. Das, Asian Pac. J. Cancer Prev., 2005, 6, 326 Search PubMed.
- S. J. Baek, K. S. Kim, J. B. Nixon, L. C. Wilson and T. E. Eling, Mol. Pharmacol., 2001, 59, 901 CAS.
- P. X. Li, J. Wong, A. Ayed, D. Ngo, A. M. Brade, C. Arrowsmith, R. C. Austin and H. J. Klamut, J. Biol. Chem., 2000, 275, 20127 CrossRef CAS.
- S. J. Baek, L. C. Wilson and T. E. Eling, Carcinogenesis, 2002, 23, 425 CrossRef CAS.
- S. H. Lee, K. Yamaguchi, J. S. Kim, T. E. Eling, S. Safe, Y. Park and S. J. Baek, Carcinogenesis, 2006, 27, 972 CAS.
- J. H. Lim, J. W. Park, D. S. Min, J. S. Chang, Y. H. Lee, Y. B. Park, K. S. Choi and T. K. Kwon, Apoptosis, 2007, 12, 411 Search PubMed.
- S. J. Baek, J. S. Kim, F. R. Jackson, T. E. Eling, M. F. McEntee and S. H. Lee, Carcinogenesis, 2004, 25, 2425 CrossRef CAS.
- P. Nair, S. Muthukkumar, S. F. Sells, S. S. Han, V. P. Sukhatme and V. M. Rangnekar, J. Biol. Chem., 1997, 272, 20131 CrossRef CAS.
- S. J. Baek, J. S. Kim, S. M. Moore, S. H. Lee, J. Martinez and T. E. Eling, Mol. Pharmacol., 2005, 67, 356 CAS.
- T. Virolle, E. D. Adamson, V. Baron, D. Birle, D. Mercola, T. Mustelin and I. de B, Nat. Cell Biol., 2001, 3, 1124 CrossRef CAS.
- W. Kaszubska, H. R. Hooft van, P. Ghersa, A. M. Raemy-Schenk, B. P. Chen, T. Hai, J. F. DeLamarter and J. Whelan, Mol. Cell Biol., 1993, 13, 7180 CAS.
- F. G. Bottone, Jr., Y. Moon, B. ston-Mills and T. E. Eling, J. Pharmacol. Exp. Ther., 2005, 315, 668 CrossRef.
- K. Tamura, B. Hua, S. Adachi, I. Guney, J. Kawauchi, M. Morioka, M. Tamamori-Adachi, Y. Tanaka, Y. Nakabeppu, M. Sunamori, J. M. Sedivy and S. Kitajima, EMBO J., 2005, 24, 2590 CrossRef CAS.
- K. N. Cho, M. Sukhthankar, S. H. Lee, J. H. Yoon and S. J. Baek, Eur. J. Cancer, 2007, 43, 2404 CrossRef CAS.
- H. U. Simon, A. Haj-Yehia and F. Levi-Schaffer, Apoptosis., 2000, 5, 415 Search PubMed.
- L. Y. Chung, T. C. Cheung, S. K. Kong, K. P. Fung, Y. M. Choy, Z. Y. Chan and T. T. Kwok, Life Sci., 2001, 68, 1207 CrossRef CAS.
- H. Babich, M. E. Krupka, H. A. Nissim and H. L. Zuckerbraun, Toxicol. in Vitro, 2005, 19, 231 CrossRef CAS.
- S. Okabe, Y. Ochiai, M. Aida, K. Park, S. J. Kim, T. Nomura, M. Suganuma and H. Fujiki, Jpn. J. Cancer Res., 1999, 90, 733 CAS.
- U. Gaur and B. B. Aggarwal, Biochem. Pharmacol., 2003, 66, 1403 CrossRef CAS.
- P. S. Steeg, Nat. Rev. Cancer, 2003, 3, 55 CrossRef CAS.
- M. Egeblad and Z. Werb, Nat. Rev. Cancer, 2002, 2, 161 CrossRef CAS.
- L. A. Liotta, U. P. Thorgeirsson and S. Garbisa, Cancer Metastasis Rev., 1982, 1, 277 CrossRef CAS.
- M. Maeda-Yamamoto, H. Kawahara, N. Tahara, K. Tsuji, Y. Hara and M. Isemura, J. Agric. Food Chem., 1999, 47, 2350 CrossRef CAS.
- B. J. El, M. H. Oak, P. Anglard and V. B. Schini-Kerth, Cardiovasc. Res., 2005, 67, 317 CrossRef.
- M. Makimura, M. Hirasawa, K. Kobayashi, J. Indo, S. Sakanaka, T. Taguchi and S. Otake, J. Periodontol., 1993, 64, 630 CAS.
- H. Oneda, M. Shiihara and K. Inouye, J. Biochem., 2003, 133, 571 CrossRef CAS.
- C. Parr, G. Davies, T. Nakamura, K. Matsumoto, M. D. Mason and W. G. Jiang, Biochem. Biophys. Res. Commun., 2001, 285, 1330 CrossRef CAS.
- H. Y. Zhou, Y. L. Pon and A. S. Wong, Endocrinology, 2007, 148, 5195 CrossRef CAS.
- U. S. Kammula, E. J. Kuntz, T. D. Francone, Z. Zeng, J. Shia, R. G. Landmann, P. B. Paty and M. R. Weiser, Cancer Lett., 2007, 248, 219 CrossRef CAS.
- R. L. Bigelow and J. A. Cardelli, Oncogene, 2006, 25, 1922 CrossRef CAS.
- D. Duhon, R. L. Bigelow, D. T. Coleman, J. J. Steffan, C. Yu, W. Langston, C. G. Kevil and J. A. Cardelli, Mol. Carcinog., 2010, 49, 739 CAS.
- I. L. Buxton, Proc. West Pharmacol. Soc., 2008, 51, 30 Search PubMed.
- K. S. Ghosh, T. K. Maiti, A. Mandal and S. Dasgupta, FEBS Lett., 2006, 580, 4703 CrossRef CAS.
- S. M. Rumjahn, M. A. Javed, N. Wong, W. E. Law and I. L. Buxton, Br. J. Cancer, 2007, 97, 1372 CrossRef CAS.
- G. F. Hu, J. Protein Chem., 1997, 16, 669 CrossRef CAS.
- M. Isemura, Y. Suzuki, K. Satoh, K. Narumi and M. Motomiya, Cell Biol. Int., 1993, 17, 559 CrossRef CAS.
- K. Ogata, N. Mukae, Y. Suzuki, K. Satoh, K. Narumi, T. Nukiwa and M. Isemura, Planta Med., 1995, 61, 472 CrossRef CAS.
- T. Kawaguchi, Curr. Drug Targets. Cardiovasc. Haematol. Disord., 2005, 5, 39 Search PubMed.
- S. Adachi, T. Nagao, S. To, A. K. Joe, M. Shimizu, R. Matsushima-Nishiwaki, O. Kozawa, H. Moriwaki, F. R. Maxfield and I. B. Weinstein, Carcinogenesis, 2008, 29, 1986 CrossRef CAS.
- J. W. Kim, A. R. Amin and D. M. Shin, Cancer Prev. Res., 2010, 3, 900 Search PubMed.
- X. Tan, D. Hu, S. Li, Y. Han, Y. Zhang and D. Zhou, Cancer Lett., 2000, 158, 1 CrossRef CAS.
- T. Kuzuhara, M. Suganuma and H. Fujiki, Cancer Lett., 2008, 261, 12 CrossRef CAS.
- G. Xie, M. Ye, Y. Wang, Y. Ni, M. Su, H. Huang, M. Qiu, A. Zhao, X. Zheng, T. Chen and W. Jia, J. Agric. Food Chem., 2009, 57, 3046 CrossRef CAS.
- D. Wang, R. Xiao, X. Hu, K. Xu, Y. Hou, Y. Zhong, J. Meng, B. Fan and L. Liu, J. Agric. Food Chem., 2010, 58, 1350 CrossRef CAS.
- T. Kuzuhara, M. Suganuma and H. Fujiki, Cancer Lett., 2008, 261, 12 CrossRef CAS.
- H. Oneda, M. Shiihara and K. Inouye, J. Biochem., 2003, 133, 571 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |