Conjugated quercetin glucuronides as bioactive metabolites and precursors of aglyconein vivo
Junji
Terao
*,
Kaeko
Murota†
and
Yoshichika
Kawai
Department of Food Science, Graduate School of Nutrition and Bioscience, The University of Tokushima, Tokushima, 770-8503, Japan. E-mail: terao@nutr.med.tokushima-u.ac.jp; Fax: +81-88-63307989; Tel: +81-88-633-7087
First published on 17th November 2010
Abstract
Quercetin is a typical anti-oxidative flavonoid ubiquitously distributed in vegetables. It is likely to act as a bioactive compound by exerting reactive oxygen species (ROS)-scavenging activity and/or binding to specific proteins such as oxidative enzymes and transcriptional factors in signal transduction pathways. Its absorption and metabolism (as well as its molecular targets) have been extensively explored from the viewpoint of its potential for disease prevention. It is known that glucuronide and/or sulfate conjugates with or without O-methylation exclusively circulate in the human bloodstream after intake of a quercetin-containing diet. We propose that glucuronide conjugates of quercetin function not only as detoxified metabolites but hydrophilic bioactive agents to various ROS-generating systems and precursors of hydrophobic aglycone. Quercetin aglycone is assumed to emerge in the target site by the action of β-glucuronidase activity under oxidative stress such as inflammation. The cardiovascular system and central nervous system seem to be the major targets of conjugated quercetin glucuronides circulating in the human bloodstream.
Introduction
Flavonoids have attracted much attention as non-nutritional food factors exerting a potential function in disease prevention and health promotion.1,2 Among them, quercetin (3,3′,4′,5,7-pentahydroxylflavone) is a flavonol-type flavonoid ubiquitously present in vegetables. Quercetin-rich vegetables such as onion and lettuce in meals are quite popular, and the daily intake of quercetin in Western countries is estimated to be 10–20 mg.3 The extensive studies using cell cultures and experimental animals have clearly indicated its anti-atherosclerotic and anti-carcinogenic effects, as well as its protective action in the central nervous system (CNS).4–11Quercetin is therefore expected to be a powerful phytochemical for disease prevention.This polyphenolic compound is known to be a strong anti-oxidant which scavenges reactive oxygen species (ROS) directly or effectively inhibits ROS-generating enzymes (e.g.xanthine oxidase, lipoxygenase).12,13 However, its catechol structure (o-dihydroxyl structure in the B-ring) leads to pro-oxidant properties by generating ROS during its oxidation process, which may result in quinone toxicity.14,15 Conversely, its pro-oxidant activity may enhance cellular defence against oxidants by stimulating the redox-regulated signal transduction pathway which leads to the gene expression of anti-oxidative enzymes such as heme oxygenase-1 (HO-1) and glutathione peroxidase (GPx).16 Furthermore, its oxidation products can act as electrophiles to bind specific proteins, resulting in the modification of their cellular functions. Quercetin is therefore expected to act as a phytochemical by exerting a wide variety of biological activities through these action mechanisms. Nevertheless, quercetin is completely converted into its conjugated metabolites by phase-II enzymes during intestinal absorption.17 Accordingly, detoxified metabolites exclusively circulate in the human bloodstream.18 Their translocation and accumulation into target sites should be clarified to estimate the effectiveness of dietary quercetin as a food factor (Fig. 1).
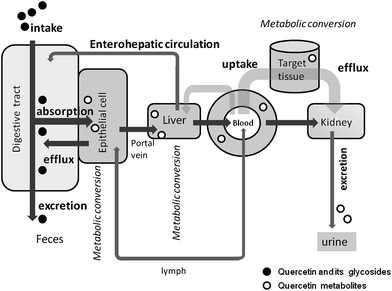 | ||
| Fig. 1 Absorption, translocation and excretion of dietary flavonoids after oral intake. Quercetin glycosides from diet are slightly absorbed via the intestinal epithelial cells, although most of them return into the digestive tract by the action of specific transporter systems. Phase II enzymes present in the intestinal epithelial cells and the liver cells facilitate their metabolic conversion to glucuronide and sulfate conjugates. Accordingly, quercetin conjugates exclusively circulate in the human bloodstream and excreted into the urine by the ingestion of quercetin glycoside-containing diet. The mechanism for the uptake and efflux in target organs has not been clarified yet. | ||
We discovered that a conjugated metabolite derived from quercetin possesses considerable anti-oxidant activity in several in vitro experiments.13 Conjugated quercetin metabolites accumulate in human atherosclerotic lesion sites, but not in normal arteries.19 An in vitro culture cell study strongly suggested that the functions of scavenger receptors of stimulated macrophages are modified by their deconjugated aglycone depending on the activity of β-glucuronidase.19 Conjugated quercetin glucuronides may therefore act as bioactive metabolites and precursors of aglyconein vivo.
In this review article, we focus on quercetin 3-O-β-D-glucuronide (Q3GlcA) as a major quercetin metabolite present in human blood and compare the biological activities of this metabolite with those of its deconjugated aglycone (Fig. 2). A possibility to release the aglycone from this conjugate during oxidative stress is also discussed.
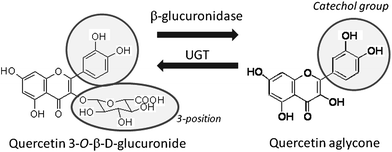 | ||
| Fig. 2 Bioconversion between Q3GlcA and its aglycone. Quercetin aglycone serves as a substrate of UGT (uridine 5′-diphosphate glucuronosyltransferase) activity in Phase-II enzyme-catalyzed detoxification. Q3GlcA can be converted to quercetin aglycone by the action of β-glucuronidase activity in inflammation. | ||
Characterization of quercetin metabolites in blood plasma
Quercetin is found in various plant food products such as tea, wine, onions, lettuce, cabbage, broccoli, beans, apples, peas, and buckwheat.3 Among them, onion is a major source of quercetin in daily foods. Quercetin in onions is present in its glucoside form, i.e., quercetin 4′-O-β-D-glucoside (Q4′G) and quercetin 3,4′-O-β-D-diglucoside (Q3,4′diG), with a smaller amount of isorhamnetin 4′-O-glucoside.20 The edible flesh of yellow onion contains these quercetin glucosides in the range 28–50 mg per 100 g.21Glucosylated quercetin in the diet is hydrolyzed to aglycone by saliva,14 but the main site for the hydrolysis of quercetin glucosides is the epithelial cells in the small intestine.22Q4′G and other quercetin monoglucosides are effectively absorbed from the intestine after hydrolysis by lactase phlorizin hydrolase (LPH) on the cell surface and/or intracellular β-glucosidase.23,24 In any case, aglycone generated is immediately metabolized during the absorption by phase-II enzymes located in intestinal cells.25Quercetin aglycone is converted into various conjugated compounds by uridine 5′-diphosphate glucuronosyltransferase (UGT) and phenol sulfotransferase (PST) and, in addition, O-methylated compounds by catechol-O-methyl transferase (COMT). In the large intestine, absorption and metabolism of quercetin is more complex because of the participation of enterobacteria.26 Quercetin glycosides other than the glucosylated form (e.g., rutin) reach the large intestine without absorption in the small intestine or without being subject to the hydrolysis to aglycone or the ring-scission to decomposition products by the action of enterobacteria.27Aglycone absorbed in large intestinal cells seems to be converted to its conjugated metabolites in a similar fashion to that in small intestinal cells. Finally, quercetin metabolites are transported into the liver, where complex conjugation reactions occur to completely detoxify quercetin.28 In this sense, quercetin is recognized to be a true xenobiotic in the human body.
Quercetin metabolites present in human blood plasma were first reported by Day et al.29 They found Q3GlcA, 3′-methyl quercetin-3-O-β-D-glucuronide and quercetin-3′-O-sulfate as major metabolites 1.5 h after the consumption of onions. Mullen et al.30 thereafter identified 23 species of quercetin metabolites from the plasma and urine 1 h after the ingestion of fried onions, in which these three conjugates were also found as major quercetin metabolites. The diphenylpropane structure (a basic skeleton of flavonoids) possesses hydrophobic properties, so quercetin aglycone is lipophilic and can therefore interact with the phospholipid bilayers of cellular and subcellular membranes.31 Recently, Fiolani et al.32 suggested that quercetin is preferentially translocated into the mitochondrial fraction of Jurkat human lymphoblast cells, indicating that the mitochondrial compartment is an intracellular target of quercetin aglycone if it is incorporated into cells. We found that quercetin aglycone passed through the cellular membranes and was metabolized by intracellular O-methylation and conjugated enzymes in mouse fibroblast cells.33Quercetin aglycone serves as a toxic substance to cultured cells by being converted to semiquinone radicals and O-quinone.14,15 We compared the effect of quercetin aglycone and Q3GlcA on cell viability using a nerve cell model and Q3GlcA was found to be much less toxic as compared with its aglycone34 even though Q3GlcA can cause quinone toxicity because of its catechol groups in the B ring. Less toxicity of Q3GlcA seems to be derived from little uptake into the cell because of its extremely high hydrophilicity due to the dissociation of acidic carboxyl group at neutral pH (Fig. 3). In addition, the lack of a free OH group at the 3-position obviously contributes to the less toxic effect of Q3GlcA because the OH group at this position is necessary to generate quinone methide from quinine.14,35Quinine methide is more electrophilic and more biologically active than o-quinine.36,37 On the other hand, Passamonti38 recently claimed that bilitranslocase is a possible system for hydrophilic flavonoids such as anthocyanins to be introduced into cells. Further studies should be developed on the specific transportation system for glucuronide quercetin conjugates to understand their mechanism of cellular uptake from a toxicological and functional viewpoint.
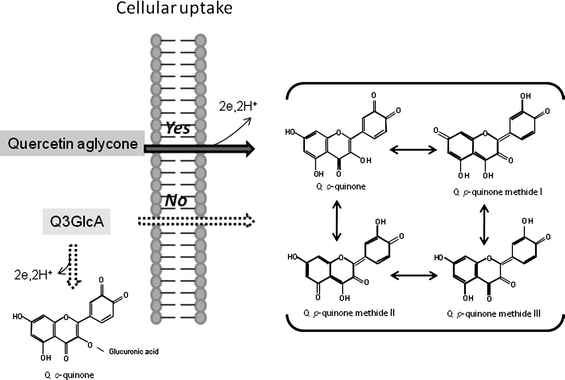 | ||
| Fig. 3 Cellular uptake of quercetin aglycone and quinine formation. Quercetin aglycone is easily incorporated into target cells through simple diffusion mechanism because of its high hydrophobicity, whereas cellular uptake of Q3GlcA hardly occurs due to its high hydrophilicity. Quercetin aglycone is oxidized to p-quinone methide by two electrons transfer reaction. Q3GlcA seems to be resistant to oxidation as compared with quercetin aglycone. In addition, Q3GlcA is unable to convert to p-quinone methide because it lacks free hydroxyl group at the 3-position. | ||
Comparison of Q3GlcA and its aglycone with respect to anti-oxidant activity in vitro
In 1999, our research team suggested that anti-oxidative compounds are involved in the plasma metabolites derived from dietary quercetin.39,40 Thereafter, we identified Q3GlcA as an anti-oxidative metabolite in rat plasma after oral administration of quercetin aglycone.41 This metabolite suppressed copper ion-induced oxidation of human low-density lipoprotein (LDL) effectively at 5 μM, because it possesses a catechol group at the B-ring, which is responsible for the chelating activity and free radical-scavenging activity.42 Then Janisch et al.43 compared the inhibitory effect of several quercetin metabolites against Cu(II)-induced LDL oxidation and they found that Q3GlcA exerts a profound inhibition at less than 2 uM. We also found that Q3GlcA (10 μM) inhibited the liposomal lipid peroxidation induced by peroxynitrite.44 Yokoyama et al.45 recently clarified that Q3GlcA at physiological level (1 μM) effectively inhibits peroxynitrite-induced nitrotyrosine formation in human serum albumin. Therefore, Q3GlcA is a preventive factor on peroxynitrite-induced oxidative damage.Shirai et al.33 demonstrated that Q3GlcA exerted anti-oxidant activity more effectively than quercetin aglycone when treated before exposure to hydrogen peroxide in mouse fibroblast 3T3 cultured cells. It can be concluded that Q3GlcA is more effective than its aglycone in the protection against oxidative stress induced by the extracellular attack of ROS, because it is stable during the incubation period even though barely incorporated into the cells. Quercetin aglycone is unstable in the medium of cultured cells, resulting in its significant decrease.
Biological activities other than anti-oxidant activity and target proteins
There are several reports concerning the inhibitory effect of quercetin glucuronides on the activities of ROS-generating enzymes such as xanthine oxidase and several types of lipoxygenases. Day et al.25 indicated that although quercetin 4′-O-β-D-glucuronide (Q4′GlcA) is a strong inhibitor of both enzymes, the effect of Q3GlcA was much weaker than that of Q4′GlcA (xanthine oxidase; Ki = 160 μM for Q3GlcA and Ki = 0.25 μM for Q4′GlcA, lipoxygenase; Ki = 60 μM for Q3GlcA and Ki = 8.4 μM for Q4′GlcA). This means that a catechol structure is not essential for the inhibition because Q4′GA loses a catechol group in the B-ring. Interestingly, we found that quercetin aglycone and Q3GlcA at 10 μM can inhibit myeloperoxidase (MPO)-mediated dityrosine formation from tyrosine in HL-60 cells.46 A docking simulation model study revealed that Q3GlcA could bind to the active site of MPO with the B-ring oriented to the heme plane in a way similar to that for the aglycone. Loke et al.47 examined the inhibitory effect of quercetin and its major metabolites on MPO-catalyzed LDL oxidation and found that glucuronidation at the 3-position still maintained a significant activity toward MPO (IC50; less than 10 μM). These phenomena suggest that Q3GlcA can act as a bioactive by protecting tissues from MPO-dependent oxidative injury during inflammation. Nicotinamide adenine dinucleotidephosphate (NADPH)-oxidase is an alternative enzyme responsible for generating ROS in the inflammation process. Neither quercetin aglycone nor Q3GlcA exhibited a remarkable inhibitory effect on NADPH-oxidase activity.48 It is likely that O-methylation is necessary to exert the inhibition because isorhamnetin exhibited a significant inhibitory effect on this enzyme activity (IC50 = 2.8 ± 1.0 μM). De Pascual-Teressa et al.49 demonstrated that Q3GlcA downregulates cyclooxyganse-2 expression in human lymphocyte at the concentration around 1 μM. Tribolo et al.50 found that Q3GlcA is able to inhibit VCAM-1 (vascular cell adhesion molecule-1) cell surface expression in human umbilical vein endotherila cells (HUVEC) at 2 μM.In human plasma, quercetin metabolites increased at ∼1 μM level after the intake of a flavonol-rich diet and then gradually decreased to nearly zero in 8 h.27,51 Our recent result shows that the concentration of Q3GlcA in human plasma was estimated to 0.26 ± 0.04 μM 1.5 h after the intake of onion (quercetin equivalent, 2.3 mg kg−1 body weight).19 This level is quite lower than the concentration at which Q3GlcA exerts effective biological activities in most cases as described above. It is likely that Q3GlcA is concentrated in the target sites where it exerts a specific function as the plasma level of Q3GlcA does not really reflect the concentration in the target sites.
As for the cellular target protein, the aryl hydrocarbon receptor (AhR) may be a target receptor for the chemopreventive effect of quercetin. Flavonoids (including quercetin) have been reported to be natural ligands for the AhR because quercetin antagonized 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced AhR transformation in a cell-free system.52 The antagonistic activity of Q3GlcA was much weaker than that of quercetin aglycone. It is therefore likely that quercetin aglycone (but not its conjugated metabolites) is a potent antagonist for the AhR and may therefore exert a preventive effect against the AhR-dependent carcinogenicity evoked by polycyclic aromatic hydrocarbons. Quercetin seems to exert a phytoestrogen-like activity similar to that observed for the isoflavone genistein, which binds to the estrogen receptor. Nevertheless, Tsuji et al.53 found that quercetin aglycone and Q3GlcA at a concentration around 10 μM could inhibit the receptor activation of nuclear factor-kappa B ligand (RANKL)-induced osteoclast differentiation, but estrogenic activity through estrogen receptors did not seem to be observed. The molecular target for quercetin may be different from that of isoflavone even though both compounds can be expected to serve as anti-osteoporotic phytochemicals. There is little information on the mechanism whereby quercetin modulates signal transduction pathways, and direct molecular targets have yet to be identified. Lee et al.54 found that quercetin was bound directly to Raf and MEK protein kinases and inhibited their phosphorylation activities in JB6 mouse epidermal cells.
Distribution of quercetin and its metabolites in tissues
De Boer et al.55 reported the distribution of quercetin in rats and pigs after feeding these animals a diet containing 0.1% or 1.0% quercetin for 11 weeks. Quercetin and quercetin metabolites were widely distributed in rat tissues, with the highest concentration in the lungs and the lowest in the brain, white fat and spleen. A similar profile was found in pigs in a short-term ingestion study (500 mg quercetin per kg body weight). However, Bieger et al.56 pointed out that long-term supplementation did not show greater accumulation of a single intake of quercetin, and the only high plasma concentrations were in the liver, small intestine and kidney. The highest concentration in the lungs of rats was not confirmed in the pig study. Although the aglycone was detected in considerable amounts, most of the aglycone could have been derived from deconjugation during the extraction procedures. Further studies are required to ascertain if the aglycone accumulates in tissues. Mullen et al.57 fed 14C-labelled Q4′G by gavage to rats (4 mg per kg body weight). They found that very little radioactivity was detected in body tissues other than the gastrointestinal tract. They therefore claimed that most of the ingested Q4′G were converted to phenolic acids before absorption into the body, and that accumulation in target tissues may be questionable. These contradictory results indicate that the degree of quercetin accumulation in body tissue is greatly dependent upon the method and amount of its administration. A study on the bioavailability of naringin (naringenin 7-O-neohesperidioside) between healthy rats and tumor-bearing rats demonstrated that the concentration of conjugated metabolites in the plasma of tumor-bearing rats was significantly lower than that of healthy rats.58 Thus, abnormal conditions (e.g., cancer) may also influence the bioavailability of flavonoids, resulting in lower accumulation in target tissues.Deconjugation accompanied by inflammation
Shimoi et al.59 first demonstrated that luteolin monoglucuronide was converted to free aglycone during inflammation using human neutrophils stimulated with ionomycin/cytochalasin B, and in rats treated with lipopolysaccharide (LPS). They found that β-glucuronidase activity in human serum from patients on hemodialysis increased significantly compared with that from healthy volunteers. We also found that LPS-stimulated macrophages enhanced β-glucuronidase activity to generate quercetin aglycone when the cells were exposed to Q3GlcA.19 It is therefore likely that the glucuronide conjugates of flavonoids can release their aglycone by enhanced β-glucuronidase activity during inflammation.Vascular targets
Recent meta-analysis of randomized controlled trials for flavonoids and flavonoid-rich food and vascular risk indicated that there was insufficient evidence to draw the conclusion about the efficacy of individual dietary flavonoids, although cacao polyphenols, soy isoflavones and tea catechins exhibited some positive effects.60 On the other hand, a rodent study suggested that oral administration of quercetin enhances the resistance against iron-ion induced plasma lipid peroxidation.40 We found that quercetin metabolites accumulated in the plasma (but not the LDL fraction) after intake of an onion-containing diet (quercetin equivalent; 68–94 mg per day) for 7 days by women.61 Plasma LDL is therefore unlikely to be the target of dietary quercetin for exerting anti-atherosclerotic effects even though oxidative modification of LDL is believed to be the initial event in atherosclerosis.62 It was confirmed that >80% of quercetin metabolites were located in the human plasma sub-fraction containing concentrated serum albumin.63 Onion consumption in the volunteers failed to enhance the ex-vivo anti-oxidant activity of the sub-fraction against free radical-induced LDL oxidation. This indicated that the amount of anti-oxidative quercetin metabolites (including Q3GlcA) bound to serum albumin was not sufficient to exert anti-oxidant activity for the protection of plasma LDL. Plasma LDL is not the target for Q3GlcA to exert its anti-oxidant activity.64Recently, Suri et al.65 demonstrated that quercetin aglycone and quercetin 3′-sulfate, but not G3GlcA, modulate cyclic GMP-dependent relaxations in pig coronary artery. Interestingly, Loke et al.66 compared the anti-atherosclerotic effect of individual dietary polyphenols, namely, quercetin, (-)-epicatechin, theaflavin, sesamin, or chlorogenic acid, in apolipoproteinE-knockout mice and found that quercetin and theaflavin selectively attenuated atherosclerosis in the mice by alleviating inflammation, improving NO bioavailability and inducing HO-1 protein in the lesions. We investigated the anti-oxidative effect of dietary quercetin on blood vessels using rabbits fed a high-cholesterol diet with 0.1% quercetin 3-O-β-glucoside for 1 month.67 Conjugated quercetin metabolites accumulated in the rabbit aorta, and the level of cholesteryl ester hydroperoxides (used as an index of lipid peroxidation) was significantly reduced, along with accumulation of conjugated quercetin metabolites. Yoshizumi et al.68 found that quercetin inhibited Src homology and collagen (Shc)- and phosphatidylinositol 3-kinase (PI3-kinase)-mediated c-Jun N-terminal kinase (JNK) activation by angiotensin II in cultured rat aortic smooth muscle cells (SMCs). Thereafter, they also clarified that quercetin aglycone and Q3GlcA prevented angiotensin II-dependent hypertrophy of vascular SMCs viainhibition of JNK and AP-1 signaling pathways.69 Ishizawa et al. recently indicated that Q3GlcA inhibits migration and proliferation by platelet-derived growth factor in vascular SMCs.70 Taken together, Q3GlcA could be a bioactive compound in the vascular system by targeting SMCs. Kawai et al.71 recently developed a monoclonal antibody that could recognize Q3GlcA, and evaluated the localization of flavonoids in tissues using immunohistochemical methods. This conjugated quercetin metabolite were found to accumulate in injured human aorta with atherosclerotic plaques.19 In particular, immunohistochemical analyses demonstrated that macrophage-derived foam cells were potential targets of dietary quercetin in the vascular system. Actually, in vitro experiments using murine macrophages showed that Q3GlcA was effectively incorporated into LPS-treated cells after deconjugation to active aglycone.19 We also found that part of the aglycone was further metabolized to O-methylated quercetin, which is responsible for the inhibition of mRNA expression of SR-A (a scavenger receptor necessary for formation of form cells). As mentioned above, inflammation is suggested to accelerate the release of cellular β-glucuronidase activity, and thus active aglycone appears to act as a modulator of the signal transduction pathway for SR-A mRNA expression.
Questions arise regarding the translocation of quercetin metabolites from the bloodstream to the arterial intima, where ROS are generated by oxidative enzymes in endothelial cells, monocyte-derived macrophages and SMCs. Mochizuki et al.72 found that the vascular permeability of Q3GlcA in human aortic endothelial cell culture inserts was dramatically increased by the stimulation of interleukin (IL-1)-alpha, an inflammatory cytokine originating from macrophages. We also found that Q3GlcA effectively passed through human umbilical vein endothelial cell monolayers in the Transwells in the presence of lysophosphatidylcholine,73 which is a product of oxidized LDL with platelet-activating factor-acetylhydrolase with potent-inflammatory and pro-atherogenic activities.74 It was also suggested that Q3GlcA is readily released from the plasma albumin fraction because the strength of its binding to serum albumin is much weaker than that of aglycone.75 These findings indicate that Q3GlcA is effectively translocated into the vascular intima if endothelial cells are subjected to oxidative stress (e.g., exposure to oxidized LDL). Therefore, circulating quercetin conjugates are likely to pass through endothelial cells to reach the intima and SMCs, where they exert their biological effects as it is or after deconjugation (Fig. 4).
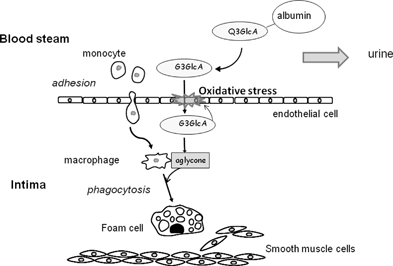 | ||
| Fig. 4 Translocation of Q3GlcA from the circulation into the arterial intima and final target sites. Q3GlcA is easily released from serum albumin-bound form and permeates into the intima, when endothelial cells are exposed to oxidative stress. Quercetin aglycone is formed from Q3GlcA by the enhanced activity of β-glucuronidase in inflammation. Quercetin aglycone is effectively incorporated into the macrophage-derived foam cells. This illustration is partly modified from the ref. 13. | ||
CNS: a possible target?
We clarified that fatty acid hydroperoxide dependent-formation of ROS in a model of cells from the CNS was effectively suppressed by Q3GlcA even though only a small amount of Q3GlcA was incorporated into the cells.76 This implies that Q3GlcA could protect nerve cells from attack by peroxidized lipids. Quercetin intake and its related flavonoids has been expected to exert a neuroprotective effect, resulting in a lower risk of CNS disorders.6,8 Nevertheless, it is uncertain if conjugated quercetin metabolites accumulate in target sites in brain tissues by crossing the blood–brain barrier. As mentioned above, animal studies demonstrated that the level of quercetin accumulated in the brain is much lower than in the liver and digestive tract. We recently investigated the effect of quercetin intake on the response to acute stress on the hypothalamic–pituitary–adrenal (HPA) axis in rats.77 Intake of the compound suppressed water immersion-restraint stress-induced increase of mRNA expression of corticotrophin-releasing factor (CRF) in the hypothalamus. This implied that the hypothalamus is a target site of dietary quercetin in the CNS. Nevertheless, there is no direct evidence on the accumulation of quercetin and/or its metabolites in this region of the brain.Monoamine oxidase-A (MAO-A) is a flavin enzyme present in the outer membrane of mitochondria. It catalyzes the oxidative deamination of serotonin to produce hydrogen peroxide and metabolized products. In the CNS, the level of MAO-A in the mitochondrial compartment of synapses is suggested to increase during major depression.78 Several studies indicate that quercetin aglycone possesses a substantial inhibitory effect on MAO-A activity.79,80 We tried to measure the efficacy of inhibition by quercetin aglycone and Q3GlcA on MAO-A activity in mitochondrial fractions of mice brains, and found that quercetin (but not Q3GlcA) had considerable activity on MAO-A inhibition.81 Instead, Q3GlcA could suppress the accumulation of hydrogen peroxide by scavenging ROS derived from the reaction of flavin enzymes (Fig. 5). Convincing data on the translocation of quercertin and/or Q3GlcA to subcellular particles in the CNS are lacking. However, mitochondria may be the ultimate target for dietary quercetin to exert its biological effect in the CNS because this compartment is suggested to concentrate quercetin aglycone to protect intracellular oxidative stress.32
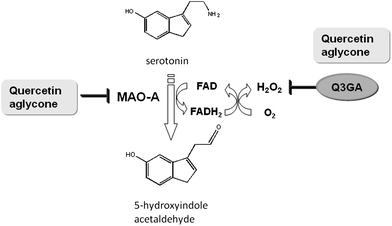 | ||
| Fig. 5 Possible role of Q3GlcA on the inhibition of the activity of mitochondrial MAO-A in the central nervous system. MAO-A (monoamine oxidase-A) is a flavin enzyme which catalyzes the oxidative deamination of serotonin to produce 5-hydroxyindole acetaldehyde and ammonia. FAD is also converted to FADH2, accompanied by the formation of superoxide anion radicals from molecular oxygen. Hydrogen peroxide is finally yielded by their disproportionation reaction. Quercetin aglycone can act as both a MAO-A inhibitor and a scavenger of superoxide anion radicals, whereas Q3GlcA serves as only a scavenger of superoxide anion radicals.81 | ||
Conclusion
Quercetin possesses powerful anti-oxidant activity due to phenolic hydroxyl groups, in particular the catechol group in the B-ring.82 Its conjugated metabolite in the bloodstream, Q3GlcA, exhibits a wide variety of physiological functions in addition to its inherent anti-oxidant activity due to the catechol group. Q3GlcA also acts as a non-toxic precursor of active aglycone. It seems to be translocated and concentrated into the target site under oxidative stress so that its physiological functions are more effective. Furthermore, aglycone is released from Q3GlcA by enhanced activity of β-glucuronidase and incorporated into cells to affect functions. One of the potential targets of dietary quercetin is the vascular system, where Q3GlcA is translocated to the ultimate targets, i.e., activated macrophages (and possibly SMCs). The CNS may be an alternative target site for dietary quercetin (although interaction at the blood–brain barrier and translocation to synaptic cells should be clarified thoroughly).In summary, Q3GlcA and other detoxified glucuronide conjugates of quercetin act as bioactive agents and potent precursors of active aglycone. The balance of the level of conjugated metabolites and its aglycone in the target site is a determinant for the effectiveness of dietary quercetin as a functional food factor for disease prevention and health promotion.
Acknowledgements
The authors acknowledge funding from the Program for the Promotion of Basic and Applied Researches for Innovations in Bio-Oriented Industry (Japan) and a Grant-in-Aid to J. T. from the Ministry of Education, Culture, Sports, Science and Technology, Japan (grant number 22380077).References
- J. A. Ross and C. M. Kasum, Annu. Rev. Nutr., 2002, 22, 19–34 CrossRef CAS.
- C. S. Fraga, Plant Phenolics and Human Health, John Wiley and Sons, Hoboken, USA, 2009 Search PubMed.
- M. G. Hertog, P. C. H. Hollman and M. B. Katan, J. Agri. Food Chem., 1992, 49, 3106–3112.
- P. C. Hollman and M. B. Katan, Arch. Toxicol., Suppl., 1998, 20, 237–248 Search PubMed.
- M. Aviram and B. Furman, Ann. N. Y. Acad. Sci., 2002, 957, 146–161 CrossRef CAS.
- K. A. Youdim, J. P. Spencer, H. Schroeter and C. Rice-evans, Biol. Chem., 2002, 383, 503–519 CrossRef CAS.
- G. Galati and P. J. O'Brien, Free Radical Biol. Med., 2004, 37, 287–303 CrossRef CAS.
- K. A. Youdin, B. Shukitt-Hale and J. A. Joseph, Free Radical Biol. Med., 2004, 37, 1683–1693 CrossRef CAS.
- S. Ramos, J. Nutr. Biochem., 2007, 18, 427–442 CrossRef CAS.
- A. Murakami, H. Ashida and J. Terao, Cancer Lett., 2008, 269, 315–325 CrossRef CAS.
- J. Terao, Y. Kawai and K. Murota, Asia Pac. J. Clin. Nutr., 2008, 17S, 291–293.
- C. Rice-Evans, N. J. Miller and G. Paganga, Free Radical Biol. Med., 1996, 20, 933–956 CrossRef CAS.
- J. Terao, Forum of Nutrition, 2009, 61, 87–94 Search PubMed.
- A. Boots, G. R. M. M. Haenen and A. Bast, Eur. J. Pharmacol., 2008, 585, 325–337 CrossRef CAS.
- D. Metodiewa, A. K. Jaiswal, N. Cenas, E. Dickancaite and J. Segura-Aguilar, Free Radical Biol. Med., 1999, 26, 107–116 CrossRef CAS.
- R. J. A. Williams, J. P. E. Spencer and C. Rice-Evans, Free Radical Biol. Med., 2004, 36, 838–849 CrossRef CAS.
- A. Scalbert and G. Williamson, J. Nutr., 2000, 130, 2073S–2085S CAS.
- G. Williamson, D. Barron, K. Shimoi and J. Terao, Free Radical Res., 2005, 39, 457–459 CrossRef CAS.
- Y. Kawai, T. Nishikawa, T. Shiba, S. Saito, K. Murota, N. Shibata, M. Kobayashi, M. Kanayama, K. Uchida and J. Terao, J. Biol. Chem., 2008, 283, 9424–9434 CrossRef CAS.
- T. Tsushida and M. Suzuki, Nippon Shokuhin Kagaku Kogaku Kaishi, 1996, 43, 642–649 CAS.
- T. Walle, A. M. Browning, L. L. Steed, S. G. Reed and U. K. Walle, J. Nutr., 2005, 135, 48–52 CAS.
- K. Murota and J. Terao, Arch. Biochem. Biophys., 2003, 417, 12–17 CrossRef CAS.
- A. J. Day, M. S. Dupont, S. Ridley, M. Rhodes, M. J. C. Rhodes, M. R. A. Morgan and G. Williamson, FEBS Lett., 1998, 436, 71–75 CrossRef CAS.
- A. J. Day, F. J. Canada, J. C. Diaz, P. A. Kroon, R. Mclauchlan, C. B. Faulds, G. W. Plumb, M. R. Morgan and G. Williamson, FEBS Lett., 2000, 468, 166–170 CrossRef CAS.
- A. J. Day, Y. Bao, M. R. A. Morgan and G. Williamson, Free Radical Biol. Med., 2000, 29, 1234–1243 CrossRef CAS.
- V. D. Bokkenheuse, C. H. L. Shacketon and J. Winter, Biochem. J., 1987, 248, 953–956.
- C. Manach, C. Morand, C. Demigne, O. Texier, F. Regerat and C. Remesy, FEBS Lett., 1997, 409, 12–16 CrossRef CAS.
- H. van de Woude, M. G. Boersma, J. Vervoort and I. M. C. M. Rietjens, Chem. Res. Toxicol., 2004, 17, 1520–1530 CrossRef.
- A. J. Day, F. Mellon, D. Barron, G. Sarrazin, M. R. A. Morgan and G. Williamson, Free Radical Res., 2001, 35, 941–952 CrossRef CAS.
- W. Mullen, A. Boitier, A. J. Stewart and A. Crozier, J. Chromatogr. A, 2004, 1058, 163–168 CrossRef CAS.
- J. Terao and M. Piskula, in Flavonoids in Health and Disease, ed, C. A. Rice-Evans , L. Packer , New York, Marcel Dekker Inc., 1997, pp. 277–294 Search PubMed.
- M. Firani, A. Guidarelli, M. Blasa, C. Azzolini, M. Candiracci, E. Piatti and O. Cantoni, J. Nutr. Biochem., 2010, 21, 397–404 CrossRef.
- M. Shirai, R. Yamanishi, J.-H. Moon, K. Murota and J. Terao, Biosci., Biotechnol., Biochem., 2002, 66, 1015–1021 CrossRef CAS.
- S. Otsuka, K. Kawabata, R. Mukai, A. Ishisaka, Y. Kawai and J. Terao, unpublished work.
- H. Awad, M. G. Boersma, J. Veroort and M. C. M. Rietjens, Arch. Biochem. Biophys., 2000, 378, 224–233 CrossRef CAS.
- J. L. Bolton and L. Shen, Carcinogenesis, 1996, 17, 925–929 CrossRef CAS.
- J. L. Bolton, E. Pisha, F. Zhang and S. Qiu, Chem. Res. Toxicol., 1998, 11, 1113–1127 CrossRef CAS.
- S. Passamonti, M. Terdoslavich, R. Franca, A. Vanzo, F. Trmer, E. Bridot, E. Petrussa and A. Vianello, Curr. Drug Metab., 2009, 10, 369–394 Search PubMed.
- N. Yamamoto, J.-H. Moon, T. Tsushida and A. Nagao, Arch. Biochem. Biophys., 1999, 372, 347–354 CrossRef CAS.
- E. L. da Silva, M. K. Piskula, N. Yamamoto, J.-H. Moon and J. Terao, FEBS Lett., 1998, 430, 405–408 CrossRef CAS.
- J. H. Moon, T. Tsushida, K. Nakahara and J. Terao, Free Radical Biol. Med., 2001, 30, 1274–1285 CrossRef CAS.
- K. Murota, Y. Mitsukuni, M. Ichikawa, T. Tsushida, S. Miyamoto and J. Terao, J. Agric. Food Chem., 2004, 52, 1907–1912 CrossRef CAS.
- M. Janisch, G. Williamson, P. Needs and G. W. Plumb, Free Radical Res., 2004, 38, 877–884 CrossRef CAS.
- M. Shirai, J.-H. Moon, T. Tsushida and J. Terao, J. Agric. Food Chem., 2001, 49, 5602–5608 CrossRef CAS.
- A. Yokoyama, H. Sakakibara, A. Crozier, Y. Kawai, A. Matsui, J. Terao, S. Kumazawa and K. Shimoi, Free Radical Res., 2009, 43, 913–921 CrossRef CAS.
- Y. Shiba, T. Kinoshita, H. Chuman, Y. Taketani, E. Takeda, Y. Kato, M. Naito, K. Kawabata, A. Ishisaka, J. Terao and Y. Kawai, Chem. Res. Toxicol., 2008, 21, 1600–1609 CrossRef CAS.
- W. M. Loke, J. M. Proudfoot, A. J. Mckinley, P. W. Needs, P. A. Kroon, J. M. Hodgson and K. D. Croft, J. Agric. Food Chem., 2008, 56, 3609–3615 CrossRef CAS.
- Y. Steffen, C. Gruber, T. Schewe and H. Sies, Arch. Biochem. Biophys., 2008, 469, 209–219 CrossRef CAS.
- S. de Pascual-Teresa, K. L. Johnson, M. S. Dupont, K. A. Oleary, P. W. Needs, L. M. Morgan, M. N. Clifford, Y. Bao and G. Williamson, J. Nutr., 2004, 134, 552–557 CAS.
- S. Tribolo, F. Lodi, C. Connor, S. Suri, V. G. Wilson, M. A. Taylor, P. W. Needs, P. A. Kroon and D. A. Hughes, Atherosclerosis, 2008, 197, 50–56 CrossRef CAS.
- P. C. Hollman, J. H. de Vries, S. D. van Leeuwen, M. J. Mengelers and M. B. Katan, Am. J. Clin. Nutr., 1995, 62, 1276–1282 CAS.
- H. Ashida, I. Fukuda, T. Yamashita and K. Kanazawa, FEBS Lett., 2000, 476, 213–217 CrossRef CAS.
- M. Tsuji, H. Yamamoto, T. Sato, Y. Mizuma, Y. Kawai, Y. Taketani, S. Kato, J. Terao, T. Inakuma and E. Takeda, J. Bone Miner. Metab., 2009, 27, 673–681 CrossRef CAS.
- K. W. Lee, N. J. Kang, Y. S. Heo, E. A. Rogozin, A. Pugliese, M. K. Hwang, G. T. Bowden, A. M. Bode, H. J. Lee and Z. Dong, Cancer Res., 2008, 68, 946–955 CrossRef CAS.
- V. C. de Boer, A. A. Dihal, H. van den Wounde, I. C. Arts, S. Wolffram, G. M. Alink, I. M. C. M. Rietjens, J. Keijer and P. C. H. Hollman, J. Nutr., 2005, 135, 1718–1725 CAS.
- J. Biegler, R. Cermak, R. Blank, V. C. J. de Boer, C. H. Hollman, J. Kamphues and S. J. Wolffram, J. Nutr., 2008, 138, 1417–1420.
- W. Mullen, J.-M. Rouanet, C. Auger, P.-L. Teissedre, S. T. Caldwell, R. C. Hartley, M. E. J. Lean, C. A. Edwards and A. J. Crozier, J. Agric. Food Chem., 2008, 56, 12127–12137 CrossRef CAS.
- M. Silberg, Gil-Izquierdo, L. Combaret, C. Remesy, A. Scalbert and C. Moran, Biomed. Pharmacother., 2006, 60, 529–535 CrossRef.
- K. Shimoi, N. Saka, R. Nozawa, M. Sato, I. Amano, T. Nakayama and N. Kinae, Drug Metab. Dispos., 2001, 29, 1521–1524 CAS.
- L. Hooper, P. K. Kroon, E. B. Rimm, J. S. Cohn, I. Harvey, K. A. Le Cornu, J. J. Ryder, W. L. Hall and A. Cassidy, Am. J. Clin. Nutr., 2008, 88, 38–50 CAS.
- J.-H. Moon, R. Nakata, S. Oshima, T. Inakuma and J. Terao, Am. J. Physiol, 2000, 279, R461–R467 CAS.
- D. Steinberg, S. Parthasarathy, T. E. Carew, J. C. Khoo and J. L. Witztum, N. Engl. J. Med., 1989, 320, 915–924 CAS.
- K. Murota, A. Hotta, H. Ido, Y. Kawai, J.-H. Moon, K. Sekido, H. Hayashi, T. Inakuma and J. Terao, J. Med. Invest., 2007, 54, 370–374 Search PubMed.
- S. B. Lotito and B. Frei, Free Radical Biol. Med., 2006, 41, 1727–1746 CrossRef CAS.
- S. Suri, X. H. Liu, S. Rayment, D. A. Hughes, P. A. Kroon, P. W. Needs, M. A. Taylor, S. Tribolo and V. G. Wilson, Br. J. Pharmacol., 2010, 159, 566–575 CrossRef CAS.
- W. M. Loke, J. M. Proudfoot, J. M. Hodgson, A. J. Mckinley, N. Hime, M. Magat, R. Stocker and K. D. Croft, Arterioscler., Thromb., Vasc. Biol., 2010, 30, 749–757 CrossRef CAS.
- C. Kamada, E. L. da Silva, M. Ohnishi-Kameyama, J.-H. Moon and J. Terao, Free Radical Res., 2005, 39, 185–194 CrossRef CAS.
- M. Yoshizumi, K. Tsuchiya, K. Kirima, M. Kyaw, Y. Suzaki and T. Tamaki, Mol. Pharmacol., 2001, 60, 656–665 CAS.
- M. Yoshizumi., K. Tsuchiya, Y. Suzaki, K. Kirima, M. Kyaw, J.-H. Moon, J. Terao and T. Tamaki, Biochem. Biophys. Res. Commun., 2002, 293, 1458–1465 CrossRef CAS.
- K. Ishizawa, Y. Izawa-Ishizawa, S. Ohnishi, Y. Motobayashi, K. Kawazoe, S. Hamano, K. Tsuchiya, S. Tomita, K. Minakuchi and T. Tamaki, J. Pharmacol. Sci., 2009, 109, 257–264 CrossRef CAS.
- Y. Kawai, A. Ishisaka, S. Saito, K. Uchida, N. Shibata, M. Kobayashi, Y. Fukuchi, M. Naito and J. Terao, Arch. Biochem. Biophys., 2008, 476, 124–132 CrossRef CAS.
- M. Mochizuki, K. Kajiya, J. Terao, K. Kaji, S. Kumazawa, T. Nakayama and K. Shimoi, BioFactors, 2004, 22, 201–204 Search PubMed.
- M. Kanda, K. Murota, A. Tokumura, Y. Kawai and J. Terao, Unpublished work.
- R. B. Gorelick, Am. J. Cardiol., 2008, 101, S34–40 CrossRef.
- A. Papadopoulou, R. J. Green and R. A. Frazier, J. Agric. Food Chem., 2005, 53, 158–163 CrossRef CAS.
- M. Shirai, Y. Kawai, R. Yamanishi, T. Kinoshita, H. Chuman and J. Terao, Free Radical Res., 2005, 40, 1047–1059.
- K. Kawabata, Y. Kawai and J. Terao, J. Nutr. Biochem., 2010, 21, 374–380 CrossRef CAS.
- J. H. Meyer, N. Ginovart, A. Boovariwala, S. Sagrati, D. Hussey, A. Garcia, T. Young, N. Praschak-Rieder, A. A. Wilson and S. Houle, Arch. Gen. Psychiatry, 2006, 63, 1209–1216 CrossRef CAS.
- F. Chimenti, F. Cottiglia, L. Bonsignore, L. Casu, M. Casu, C. Floris, D. Secci, A. Bolasco, P. Chimenti, A. Granese, O. Befani, P. Turini, S. Alcaro, F. Ortuso, G. Trombetta, A. Loizzo and I. Guarino, J. Nat. Prod., 2006, 69, 945–949 CrossRef CAS.
- L. Saaby, H. B. Rasmussen and A. K. Jäger, J. Ethnopharmacol., 2009, 121, 178–181 CrossRef CAS.
- S. Yoshino, A. Hara, H. Sakakibara, K. Kawabata, A. Tokumura, A. Ishisaka, Y. Kawai and J. Terao, Nutrition, 2011 DOI:10.1016/j.nut.2010.09.002.
- W. Bors, W. Heller, C. Michel and M. Saran, Methods in Enzymology, San Diego, Academic Press, 1990, vol. 186, pp343–355 Search PubMed.
Footnote |
| † Present address: Department of Life Science, School of Science and Engineering, Kinki University, Osaka 577-8502, Japan |
| This journal is © The Royal Society of Chemistry 2011 |