Polyfluoroalkyl compounds in the aquatic environment: a review of their occurrence and fate
Lutz
Ahrens
*
Department for Environmental Chemistry, Institute for Coastal Research, GKSS Research Centre Geesthacht, D-21502, Geesthacht, Germany
First published on 28th October 2010
Abstract
The occurrence and fate of polyfluoroalkyl compounds (PFCs) in the aquatic environment has been recognized as one of the emerging issues in environmental chemistry. PFCs comprise a diverse group of chemicals that are widely used as processing additives during fluoropolymer production and as surfactants in consumer applications for over 50 years. PFCs are known to be persistent, bioaccumulative and have possible adverse effects on humans and wildlife. As a result, perfluorooctane sulfonate (PFOS) has been added to the persistent organic pollutants (POPs) list of the Stockholm Convention in May 2009. However, their homologues, neutral precursor compounds and new PFCs classes continue to be produced. In general, several PFCs from different classes have been detected ubiquitously in the aqueous environment while the concentrations usually range between pg and ng per litre for individual compounds. Sources of PFCs into the aqueous environment are both point sources (e.g., wastewater treatment plant effluents) and nonpoint sources (e.g., surface runoff). The detected congener composition in environmental samples depends on their physicochemical characteristics and may provide information to their sources and transport pathways. However, the dominant transport pathways of individual PFCs to remote regions have not been conclusively characterised to date. The objective of this article is to give an overview on existing knowledge of the occurrence, fate and processes of PFCs in the aquatic environment. Finally, this article identifies knowledge gaps, presents conclusions and recommendations for future work.
 Lutz Ahrens | Lutz Ahrens obtained his diploma and PhD in environment chemistry at Leuphana University of Lüneburg, Germany. He carried out research in the transportation mechanisms of polyfluoroalkyl compounds (PFCs) in water and their bioaccumulation in biota at the Helmholtz-Zentrum Geesthacht, Germany, meanwhile working at the National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Japan, and Norwegian Institute for Air Research (NILU), Tromsø, Norway. He is currently carrying out research at Environment Canada. Current interests include the investigation of sources, fate and transport of PFCs and other emerging contaminants in the environment. |
Environmental impactThis is a critical review about the occurrence and fate of polyfluoroalkyl compounds (PFCs) in the aquatic environment. The physical transport and multimedia partitioning of PFCs depends on their physicochemical properties which vary depending on their chain length and functional group. Key loss processes and deposition, the relationship between sources and aqueous environment concentrations, partitioning behaviour and transport mechanism are discussed to lead to a better understanding of the global geochemical cycle and fate processes of PFCs in the aqueous environment. |
Introduction
In recent years, the occurrence and fate of polyfluoroalkyl compounds (PFCs) in the aquatic environment has been recognized as one of the emerging issues in environmental chemistry. PFCs comprise a diverse group of chemicals that are widely used as processing additives during fluoropolymer production and as surfactants in consumer applications, including surface coatings for textiles, furniture, and paper products for over 50 years.1In general, neutral PFCs like perfluoroalkyl sulfonamides (FASAs), perfluoroalkyl sulfonamidoethanols (FASEs) and fluorotelomer alcohols (FTOHs) are less water-soluble and more volatile than ionic PFCs. Once released in the environment, neutral PFCs can be (bio)degraded in the atmosphere or under aerobic conditions to perfluoroalkyl carboxylates (PFCAs) and perfluoroalkyl sulfonates (PFSAs).2–4 PFCAs and PFSAs are persistent against typical environmental degradation processes and have been found ubiquitously in water,5 air,6food,7 wildlife8 and humans.9 PFSAs and longer chain PFCAs are known to be bioaccumulative10,11 and have possible adverse effects on humans and wildlife.12 As a result, perfluorooctane sulfonate (PFOS) has been added to the persistent organic pollutants (POPs) list of the Stockholm Convention in May 2009 resulting in global restriction of its production.13 However, their homologues, neutral precursor compounds and new PFCs classes (e.g., perfluoroalkyl phosphonates (PAPs)) continue to be produced.
The global fate and transport pathways of PFCs have not been conclusively characterized to date. Two main hypotheses were proposed for the global transport of PFCs. First, neutral, volatile precursor compounds could undergo long-range atmospheric transport and be degraded in remote regions,2,3,14 or second, ionic PFCs could be transported directly by oceanic currents or by means of sea spray.15,16 Modelling studies mostly identify the ocean currents as the dominant transport pathway to remote regions,15,17 however, measurements of PFCs in the aqueous environment are limited and further research is needed.
This article focuses on PFCs in the aqueous environment, with the objectives, as follows:
1. Introducing the different groups of PFCs and their analytical methods for the aqueous phase.
2. Discussing potential sources for PFCs into the aqueous environment.
3. Summarising data on the typical aqueous environment concentrations.
4. Discussing the fate of PFCs after their release in the aqueous environment.
5. Evaluating their mechanisms for long-range transport.
6. Discussing the current challenges in fate and global geochemical cycle processes.
The objective of this article is therefore to give an overview on existing knowledge of the occurrence, fate and processes of PFCs in the aqueous environment. Finally, this article identifies knowledge gaps, and presents conclusions and recommendations for future work.
Analytes and analytical methodologies
PFCs comprise a wide range of different substances, consisting of a hydrophilic functional group and a lipophilic fluorinated chain which can vary in chain length. The lipophilic part is fully or partially fluorinated and can be linear or branched. The most investigated compounds are PFOS and perfluorooctanoate (PFOA). But there are several hundreds of PFCs, which can be divided into the ionic and neutral PFCs (see Table 1). The unique characteristics of PFCs, low concentration levels, different matrices and procedure and instrumental blank contamination are all analytical challenges with determining PFCs in the aqueous environment. Yamashita and co-workers eliminated sources of procedure and instrumental blank contaminations to detect PFCs for the first time in pg per litre range in oceanic waters.5 Several comprehensive studies have already been published focusing on sample pre-treatment, extraction, clean-up strategies and instrumental determination.18–20 The current analytical methodologies to determine PFCs in aqueous media is described in the following.| Compound groups | Acronym | Chemical structure | Typical PFCs |
|---|---|---|---|
| Ionic PFCs | |||
| Perfluoroalkyl sulfonates | PFSAs |
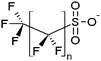
|
n = 3–9 |
| Perfluoroalkyl sulfinates | PFSiAs |
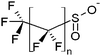
|
n = 5, 7, 9 |
| x:2 Fluorotelomer sulfonates | x:2 FTS |
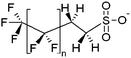
|
n = 5, 7, 9 |
| Perfluoroalkyl carboxylates | PFCAs |

|
n = 1–17 |
| Perfluoroalkyl phosphonates | PFPAs |

|
n = 5, 7, 9 |
| Fluorotelomer carboxylates | x:2 FTCA |
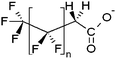
|
n = 5, 7, 9 |
| Fluorotelomer unsaturated carboxylates | x:2 FTUCA |
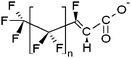
|
n = 4, 6, 8 |
| Neutral PFCs | |||
| x:2 Fluorotelomer olefins | x:2 FTolefin |
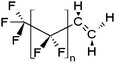
|
n = 5, 7, 9, 11 |
| x:2 Fluorotelomer alkohols | x:2 FTOH |
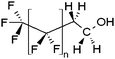
|
n = 3, 5, 7, 9, 11 |
| x:2 Fluorotelomer acrylates | x:2 FTA |
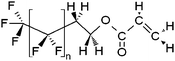
|
n = 5, 7, 9 |
| x:2 Fluorotelomer aldehydes | x:2 FTAL |

|
n = 7 |
| Perfluoroalkyl sulfonamides | FASAs |
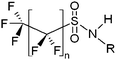
|
n = 7, R = H |
| n = 7, R = CH3 | |||
| n = 7, R = C2H5 | |||
| n = 3, R = CH3 | |||
| Perfluoroalkyl sulfonamidoethanols | FASEs |

|
n = 7, R = CH3 |
| n = 7, R = C2H5 | |||
| n = 3, R = CH3 | |||
| Perfluoroalkyl sulfonamidoacetic acids | FASAAs |
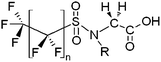
|
n = 7, R = H |
| n = 7, R = CH3 | |||
| n = 7, R = C2H5 | |||
The collection of water samples for PFC analysis is critical because the sampling method (e.g., bottle sampler, ship inlet system, rosette-type sampler), sampling period (e.g., grab sample, flow-proportional composite sample), sampling container (e.g., glass, polypropylene (PP), stainless steel) and sampling depth can have a high influence on the results. Furthermore, polyfluorinated material (e.g., polytetrafluoroethylene (PTFE)) or incorrect handling can easily contaminate the sample. A comparison of two sampling techniques (i.e., bottle sampler and ship inlet system) had shown no significant differences in seawater samples from the Atlantic Ocean,21 however, further comparison of sampling methods are necessary. The sampling depth also has an influence on the results. For example, the vertical profiles in the marine water column were associated with the global ocean circulation system22 and an enrichment by a factor of 24–109 of PFOS was found in the surface microlayer (50 μL thickness) compared to the corresponding subsurface water layer (>30 cm depth).23 The sampling and storage container could be critical because it was observed that some PFCs blank contamination was due to adsorption on glass24 or PP.25 In addition, volatile, neutral PFCs (e.g., FTOHs, FASAs) could evaporate25 or degrade to ionic PFCs (e.g., PFCAs, PFSAs).4
A filtration step is necessary for water samples with a high content of suspended particulate matter (SPM) to avoid blocking of the solid phase extraction (SPE) cartridges. In addition, a separate analysis of the dissolved phase and particulate phase enable examination of the partitioning behaviour of PFCs.26 However, during the filtration, PFCs can be adsorbing to the filtration equipment. PFCs in the dissolved phase can also adsorb to the filter material (e.g., glass fibre filter (GFF) or syringe nylon membrane filter) and the filtration equipment may also be a source for blank contamination.26,27 Before extraction, the samples should be spiked with mass-labelled surrogate standard mixtures at concentrations close to the environmental level to allow for corrections of losses during extraction, extract cleanup, and matrix effects during analysis. The most common extraction method for aqueous samples is the SPE firstly described by Moody and Field.28 The SPE method was further optimised by Taniyasu and coworkers to determine a wide range of PFCs including short and long chain PFCs.25,29,30 Alternatively, liquid–liquid extraction (LLE) was used without prior filtration, but this method was limited to the longer chain PFCs (C ≥ 8).31 Before instrumental analysis, an injection standard (InjS) should be added for quality control. To improve the method detection limits, the sample amount for the extraction can be increased or the volume of the final extract can be reduced.32 However, a higher enrichment factor of the samples can increase the instrumental matrix effects and an additional cleanup step might be necessary. An appropriate clean-up method has been described by Powley et al. using ENVI-Carb™.33 Large-volume injection can be used alternatively to analyse PFCs directly without sample pre-treatment.34
The most frequently used instrument for the measurement of PFCs is the high performance liquid chromatography coupled with a tandem mass spectrometry operated in a negative electrospray mode (HPLC/(–)ESI-MS/MS) or high resolution time-of-flight (TOF)-MS.20,35 Modifications of the instrument might be necessary to minimise contact with fluorine-containing materials.36 For example non-fluorinated tubing and seals can be used and a scavenger cartridge can be installed between the pump and injector to trap contaminants originating from the degasser, connecting tubes, and the mobile phase. Unless the samples are analysed immediately, adsorption to glass vials may occur. Better peak shape and sensitivity was observed if the sample was injected in a solution of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 methanol/water rather than 100% methanol.37 The effect of matrix inducted signal suppressions during the instrumental analysis was observed for some compounds depending on the extraction volume and the sample type.18,38 Recently a method for the determination of the total fluorine (TF), followed by fractionation of the samples to determine inorganic fluorine and organofluorine separately, was developed.39 The method was based on combustion ion chromatography (CIC) and further developed to analyse TF in low microgram per litre levels in aqueous matrices by reducing high background levels. This method can help identify the content of unknown PFCs in the sample.
:20 methanol/water rather than 100% methanol.37 The effect of matrix inducted signal suppressions during the instrumental analysis was observed for some compounds depending on the extraction volume and the sample type.18,38 Recently a method for the determination of the total fluorine (TF), followed by fractionation of the samples to determine inorganic fluorine and organofluorine separately, was developed.39 The method was based on combustion ion chromatography (CIC) and further developed to analyse TF in low microgram per litre levels in aqueous matrices by reducing high background levels. This method can help identify the content of unknown PFCs in the sample.
The purity of the standards has improved and more mass-labelled standards are available to compensate for signal enhancement/suppression or losses during sample preparation, resulting in significant improvement in results.18,38 If the PFC standard is a salt (e.g., PFOS potassium salt), then the quantification results should be calculated for the corresponding acid. In the case of strong matrix effects the standard addition method can be an alternative calibration option.38 Recently, the quantification of branched isomers has become more important because of their toxic relevance40,41 and the PFC isomer pattern can be used to identify the dominant source from either historical releases (i.e. electrochemical fluorination (ECF) fluorochemicals (mixture of isomers)) or current releases (i.e. telomer-derived products (only linear)).42
Inter-laboratory comparison studies show an improvement of the limits of detection, accuracy, precision, and robustness for the analysis of PFCs.18,38,43 But the control of blank contamination and matrix effects will be still an important issue for the future. Matrix effects can be reduced by using more sensitive instruments upon dilution. However, comparing results to published data of PFC levels in the aqueous environment should be done with caution, because of the use of many different sampling methods, pre-treatments and instruments chosen for analysis. Furthermore, different matrices (i.e., precipitation, groundwater, seawater, river water, lake water, wastewater) with different characteristics (e.g., content of SPM, pH value, organic carbon) can also influence the results. Inter-laboratory comparison studies, international standard methods (e.g., ISO 25101) and the use of certified reference materials may help to improve the quality and comparability of the results.30,38 Recently, a document was published which provide advices on the analysis of PFCs in water, including sampling, pre-treatment and instrumental analysis.44 The analytical challenges for the future are to optimise or develop new methods to determine branched isomers of PFCs and new PFC classes (e.g., PAPs) in the aqueous environment. In addition, the development of passive samplers for the aqueous environment could also be an interesting challenge for the future.
Sources and origins
Longer chain PFCs (C ≥ 3) are man-made chemicals, whereas trifluoroacetic acid (TFA) also has natural sources.45 However, it might be possible that volcano activities are a natural source for longer chain PFCs to the atmosphere. They can enter the aqueous environment during product manufacturing processes, supply chains, product use, and disposal of various industrial and consumer products.46 The PFC-based products were used for carpets (14–48%), apparel (43–48%), paper and packaging products (15–28%), aqueous film forming foam (AFFF) (6–16%) and other performance chemicals (8–20%).46,47 Dinglasan-Panlilio and Mabury found that 0.04–3.8% (dry mass basis) of residual fluoro alcohols can be found in commercial products containing PFCs.48Historic emissions are estimated to be 6800–45250 t for perfluorooctylsulfonyl fluoride (POSF) (1972–2002), from which the majority was estimated to have been released to the aqueous environment (up to 45000 t) and only a small amount into the air (up to 235 t).46 The emissions for PFCAs range between 3200 and 7300 t (1951–2004), which is lower than POSF-based production.17 However, the emissions of some PFCs (e.g., PFOS and PFOA) are assumed to have decreased because of restrictions and regulations. For example, the 3M Company voluntarily phased out the production of POSF in 2002, the US Environmental Protection Agency (US EPA) launched a voluntary stewardship program to reduce PFOA and related chemicals from facility emissions49 and the European Union (EU) prohibited the general use of PFOS and their derivates in June 2008.50 However, a variety of related PFCs are still being produced by other manufacturers17 and PFC precursors can be degraded to persistent degradation products (e.g., PFCAs and PFSAs). In addition, after they distribute in the environment PFCs can be re-emitted (e.g., from ice or sediment), which is a process that must be considered in order to understand the geochemical cycle of PFCs on a global scale.
Sources of PFCs in the aqueous environment can be generally grouped into point and nonpoint sources based on their origin. Point and nonpoint sources into the aqueous environment are industrial or municipal wastewater treatment plants (WWTPs), landfill leachate, dry or wet atmospheric deposition, and soil or street surface runoff (Table 2).28,51–54 Industrial or municipal WWTPs are mostly discussed as point sources for PFCs in the literature.55–60WWTP mass flow studies found similar or higher PFC concentrations in the effluent in comparison to the influent, which indicate that conventional WWTPs are not effective for removal of PFCs and biodegradation of precursor compounds could lead to increasing concentrations of PFCAs and PFSAs.55,56 However, the concentration levels and PFC composition profiles in WWTP effluent depends on the cleaning treatment system.55,56 The ∑PFC concentrations in WWTP effluents were about 5–10 times higher (30.5–266.3 ng L−1) than in river water samples (7.6–26.4 ng L−1).57 The major contaminants in WWTP effluents were perfluorobutane sulfonate (PFBS), PFOS and the C4–C9 PFCAs with no large seasonal variations.57–60 Interestingly, the concentration of PFBS was higher than PFOS in nine WWTPs along the River Elbe, Germany,57 which might stem from the substitution of the C8-based compounds by C4-based compounds after the voluntary phase-out of POSF-based production.61 Industrial influenced WWTPs have significantly higher PFC concentrations than WWTP which are only influenced by domestic wastewater.56,62 For example, extremely high ∑PFC concentrations were found in semiconductor, electronic, and optoelectronic industrial wastewaters in Taiwan in the mg per litre range.63 Generally, the per capita discharge was estimated to be 57 μg day−1 person−1 for PFOS and 12 μg day−1 person−1 for PFOA in the Glatt Valley Watershed, Switzerland.58 The total ∑PFCs discharged ranged from tens to hundreds of grams per day.57,58,64 The ∑PFC concentrations in the kilograms per day range were found in one industrial WWTP located at the River Rhine with perfluorobutanoate (PFBA) and PFBS as the major contaminants.65
| Country | PFOS | PFOA | Other PFCs | Source of PFCs | Reference |
|---|---|---|---|---|---|
| a n.d. = not detected; n.a. = not analysed; <MQL = below method quantitation limit. b mean values. | |||||
| Snow | |||||
| USA | <MQL–1.93 | <MQL–20 | PFHxS; C7, C9–C12 PFCA; PFOSA; 6:2, 8:2 FTS |
atmospheric deposition | Kim and Kannan 2007 |
| Canada | 0.002–0.09 b | 0.01–0.15 b | C9–C11 PFCA | atmospheric deposition | Young et al. 2007 |
| Precipitation | |||||
| Canada | 0.59 b | n.d. | PFHxS; 8:2, 10:2 FTCA; 8:2, 10:2 FTUCA |
degradation from volatile precursors | Loewen et al. 2005 |
| North America | n.a. | <MQL–89 | C2–C7, C9–C12 PFCA; 8:2, 10:2 FTCA; 8:2, 10:2 FTUCA |
degradation from volatile FTOHs | Scott et al. 2006 |
| Japan | 0.13–1.0 | 1.0–3.8 | C2–C7, C9–C12 PFCA; 8:2 FTCA; 6:2, 8:2, 10:2 FTUCA; PFOSA, EtFOSAA |
degradation from volatile precursors | Taniyasu et al. 2008 |
| Tap water | |||||
| Japan | 0.1–50.9 | n.a. | n.a. | Tama river | Harada et al. 2003 |
| Japan | n.d.–12 b | 0.12–40 | n.a. | river water | Saito et al. 2004 |
| Germany | n.d.–22 | n.d.–519 | PFBS; C4–C7 PFCA | runoff from contaminated soil | Skutlarek et al. 2006 |
| Italy | 6.2–9.7 | 1.0–2.9 | C7, C9–C12PFCA | Lake Maggiore | Loos et al. 2007 |
| Groundwater | |||||
| USA | n.a. | n.d.–6570000 | PFHxA; PFHpA | AFFFs | Moody and Field 1999 |
| USA | 19–87 | n.d.–18 | PFHxS; PFDS; C6, C7, C10PFCA; PFOSA; EtFOSAA | infiltration from overlying urban stream | Plumlee et al. 2008 |
| Surface runoff | |||||
| USA | <MQL–15 | 0.51–29 | PFHxS; C7, C9–C12 PFCA; PFOSA; 6:2, 8:2 FTS |
surface, rain | Kim and Kannan, 2007 |
| Japan | 2.9–12 | n.d.–174 | C7, C9–C12, C14PFCA; PFOSA | atmospheric deposition, dust | Murakami et al. 2009 |
| River water | |||||
| USA | 17–144 | <MQL–598 | n.a. | fluorochemical manufacturing facility | Hansen et al. 2002 |
| Japan | 0.24–37 | 0.1–456 | n.a. | various sources | Saito et al. 2004 |
| Germany | n.d.–193 | n.d.–3640 | PFBS; C4–C7 PFCA | runoff from contaminated soil | Skutlarek et al., 2006 |
| China | 0.15–99 | 0.85–260 | PFBS; PFHxS; C6, C7, C9–C11 PFCA; PFOSA | industrial/municipal wastewater effluent | So et al. 2007 |
| Europe | n.a. | <MQL–200 | C6, C7, C9PFCA | various sources | McLachlan et al. 2007 |
| Germany | 0.18–8.2 | 2.9–12.5 | 18 other PFCs | various sources | Ahrens et al. 2009 |
| Lake water | |||||
| Canadian | 0.9–57 | 0.5–16 | PFHxS; PFDS; C7, C9–C12 PFCA; | atmosphere, airport wastewater | Stock et al. 2007 |
| Arctic | 8:2, 10:2 FTUCA |
||||
| Lake Victoria | <MQL–2.5 | 0.4–12 | n.a. | industrial/municipal wastewater effluent | Orata et al. 2009 |
| Waste water effluent | |||||
| USA | 3–68 | 58–1050 | PFHxS, C9–C11 PFCA, 8:2 FTCA, 8:2 FTUCA |
industrial, domestic, and commercial influents | Sinclair and Kannan 2006 |
| USA | 20–187 | 12–185 | PFHxS, PFDS, C6, C7, C9, C10 PFCA 6:2 FTS, PFOSA, EtFOSAA |
industrial, domestic, and commercial influents | Plumlee et al. 2008 |
| Landfill effluent | |||||
| Finland, Norway | 30–187 | 91–516 | PFBS; PFHxS; C6, C9PFCA; PFOSA | landfill | Kallenborn et al. 2004 |
| Denmark | <MQL–3.8 | <MQL–5.8 | PFHxS | landfill | Bossi et al. 2008 |
| Germany | 0.01–235 | <0.40–926 | 35 other PFCs | landfill | Busch et al. 2010 |
Other potential point sources into the aqueous environment are landfill leachates.66 The leachates are usually purified in a special treatment process. Some of these treatment systems (e.g., active carbon or membrane filtration) are able to remove contaminations of PFCs efficiently from wastewater,67 but the main problem is with the disposal of the sludge and filter (e.g., burned in incinerators or deposited in landfill sites). Furthermore, the leachate can also enter the groundwater directly without undergoing any cleaning processes. High ∑PFC contamination were reported in landfill effluents (199–1537 ng L−1), but due to the low mass flow of the leachate, the total discharge was estimated to be only ∼1% compared to that of the WWTP effluents in Germany.67 However, further studies are necessary which focus on the correlation of the individual PFC concentration with the deposited waste type and also implementation of long-term monitoring programs.
The identification of nonpoint sources is more difficult than of point sources. The presence of PFC precursor compounds in the gaseous and particulate phases of the atmosphere6,68 and in precipitation29,69 indicate that dry and/or wet atmospheric deposition are potential nonpoint sources for PFCs. Wet deposition rate for ∑PFCAs were estimated to be 540–12471 ng m−2 with TFA as the dominating compound.69 The decreasing PFOS concentration in Arctic ringed seals (Phoca hispida) was explained by the reduced emissions of POSF-based compounds into the atmosphere.70
Another potential nonpoint source is soil or street surface runoff which is influenced by precipitation. Surface runoff was investigated in Albany, NY, USA, but the mass balance analysis performed on an urban lake suggests that the surface runoff is not the dominant input pathway into the lake.51 High contamination in the Möhne river was believed to originated from PFC contaminated soil fertilizer applied on agriculture lands which entered the river system by surface runoff,71 and one study identified street runoff as a potential source for PFCAs into the aqueous environment.72 Other potential sources for PFCs are AFFF spill (e.g., fire-training locations or airports)73,74 or contamination of groundwater by infiltration.75
Overall, whether point source or nonpoint sources dominate depended on the area. However, the occurrence of PFCs in the aqueous environment is highly influenced by urban activities, which is shown by a positive correlation of PFCs concentrations with population density.58,76 The detected congener composition in environmental samples may provide information to their sources and transport pathways.77,78 For example, when even-carbon PFCA patterns are higher in comparison to the odd-carbon PFCA patterns, this could indicate degradation of their precursors originating from atmospheric transport.78 The presence of the branched isomers could provide information of either ECF production or telomerisation process.42 However, the pattern could be altered as a result of different characteristics of the linear and branched isomers (e.g., different partitioning or uptake/eliminate rates in biota).40,41 Finally, PFCs are transported predominately by the rivers into the marine environment.79
Occurrence of PFCs in the aquatic environment
PFCs were ubiquitously found in the aqueous environment. About 40 PFCs were detected while the concentrations generally ranged between pg and ng per litre for individual compounds. In the following paragraphs, the occurrence of PFCs in snow, precipitation, tap water, groundwater, river water, lake water and seawater is described.Snowfall was identified as a significant pathway for PFCs into lakes in Albany, NY, USA.51 Another study investigated PFCs in ice caps from the Canadian Arctic to study seasonal cycles, temporal trends and atmospheric fluxes.80 The concentrations ranged between tens of pg to hundreds pg per litres, with maximum concentrations in spring to summer. The concentration of PFOS decreased significantly between 1996 and 2005, while no trend was observed for the PFCAs. The presence of ionic PFCs in Arctic snow indicates that wet deposition may be a source for PFCs in remote regions.
TFA was investigated in precipitation in several studies.29,81 The sources of this short chain PFC are photochemical degradation of chlorofluorocarbons (CFCs) and direct anthropogenic and natural emissions. Short and long chained PFCAs (C2–C12) and their potential precursor compounds fluorotelomer carboxylates (FTCAs) and fluorotelomer unsaturated carboxylates (FTUCAs) were detected in precipitation from nine sampling sites in North America.69 Interestingly, high PFOA concentrations correlated with air masses coming from urban areas. Dominated compounds were the short chain TFA and perfluoropropanoate (PFPrA) with maximum concentrations of 75.9 ng L−1 and 10.3 ng L−1, respectively, at two locations in Japan.29 Potential precursor compounds of PFCAs were detected in precipitation from Kyoto, Japan82 and Winnipeg, Canada.52 These results suggest that neutral PFCs can remove through oxidation and wet deposition from the atmosphere.
Tap water has been previously shown to be an exposure route of PFCs for humans.83 PFOS was detected in drinking water from Japan with a concentration of less than 4 ng L−1, except for one sampling site that had the maximum concentration of 50.9 ng L−1.84 The origin of the contamination was possibly the Tama River, which was contaminated with PFOS. A correlation of the PFC concentrations in drinking water with the concentrations in lake Maggiore, Italy, indicate an insufficient performance of the waterworks system for PFCs in this region.85 High PFC concentrations were found in drinking water in the Ruhr area, Germany, with a maximum concentration of 598 ng L−1 for the ∑PFCs.71 The high concentration originated from PFC contaminated soil, which reached the drinking water by infiltration into the groundwater.
C6–C8 PFAC were detected in groundwater ranging from 125–7090 μg L−1 at two fire-training locations in the USA, which is the first study of PFCs in aqueous samples.28 These extremely high concentrations can be explained by using of AFFFs, which contained high levels of PFCs. But the high concentrations measured even after 7–10 years of inactivity indicate the high persistency of PFCs in the aqueous environment. ∑PFC concentrations ranged from 20–150 ng L−1, with PFOS and PFOA as the dominated compounds, in groundwater in California, USA.75 The overlying urban stream possibly contaminated the groundwater through infiltration.
PFC concentrations measured in the surface water of the rivers were reported in several studies. A global comparison of the PFC congener pattern in river waters is given in Fig. 1. Generally, the C8-PFCs, PFOS and PFOA, were the dominant compounds, except in the lower Rhine, Germany, where the C4-PFCs, PFBS and PFBA, dominated (24% and 60%, respectively), and in the Ganges River, India, where perfluorohexanoate (PFHxA) dominated (58%).65
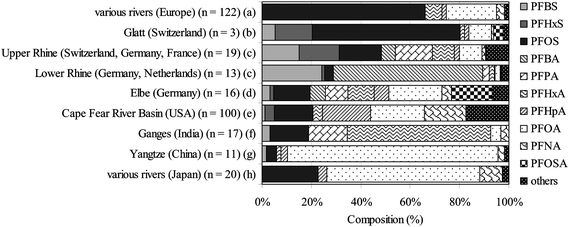 | ||
| Fig. 1 Global comparison of the PFC pattern in river waters ((a),75 (b),57 (c),64 (d),26 (e),85 (f),86 (g),87 (h)58). | ||
In a comprehensive study for PFOS and PFOA in river samples all over Japan, the concentration of PFOA was generally higher than PFOS, especially at contaminated sampling sites. Two studies on PFC concentrations in European rivers showed the highest concentrations for PFOA with 200 ng L−1 in the river Po, Italy,79 and 1371 ng L−1 in the river Krka, Slovenia.76 Overall, these studies show the widespread occurrence of PFCs in Japanese and European rivers and the large geographical differences in their levels.
PFOS concentrations of up to 2270000 ng L−1 were detected in Toronto, Canada, originating from AFFF spill.86 High PFOS and PFOA concentrations were also found in the Tennessee River, USA, originating from a fluorochemical manufacturing facility.87WWTP effluents were identified as local sources for PFCs in the Cape Fear River Basin in North Carolina, USA,88 in the Glatt Valley Watershed in Switzerland,58 river Rhine in Germany,65 and in several rivers in Japan.59 In summary, sources for PFCs into the rivers were identified to be from AFFF spills, fluorochemical manufacturing effluents, WWTP effluents and runoff, while the spatial distribution and the composition profile of individual PFCs can be used to identify the origin of the contamination.
The dominating PFCs in the Great Lakes were PFOS (21–70 ng L−1) and PFOA (27–50 ng L−1).89 In addition, some precursor compounds (e.g., perfluorooctane sulfonamide (PFOSA)) were detected. PFC concentrations in lake water from two other studies ranged from mid pg to mid ng per litres in the Canadian Arctic54 and Lake Victoria, Kenya.90 In general, the presence of PFCs in remote lakes indicates the ubiquitous distribution of PFCs in the aqueous environment.
An overview on open-ocean and coastal seawater concentrations is shown in Fig. 2. Concentrations are usually around tens of pg to a few ng per litres, depending on the location and the compound. The concentrations were approximately two orders of magnitude higher in the coastal areas in comparison to the open-ocean.
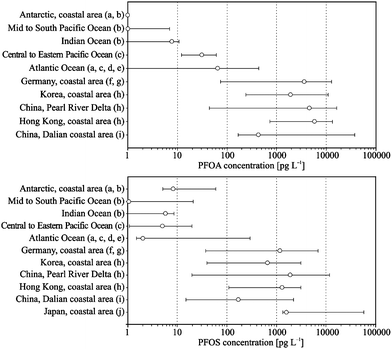 | ||
| Fig. 2 Concentrations (minimum, maximum, median (circles)) of PFOA and PFOS in seawater in the open-ocean and the coastal area in pg L−1 ((a),92 (b),93 (c),5 (d),94 (e),21 (f),95 (g),96 (h),97 (i),23 (j)98). Note: Concentrations below the method quantification limit are given as one-half of the method detection limit. | ||
Taniyasu et al. (2003) investigated PFSAs in coastal seawater around Japan.91 Maximum PFOS concentration was found in the Tokyo Bay at 59 ng L−1, while the other PFSAs were below the method detection limit (MDL). The PFC concentrations in the Pearl River Delta (China), coastal area of Hong Kong and Korea were about a few to tens of ng per litre range except at one sampling location close to the urbanized and industrial city of Seoul, South Korea, with a maximum concentration of 730 ng L−1 for PFOS.92 Similar concentration levels were observed for PFOS and PFOA in the coastal areas of Dalian, China.23 Another study investigated the occurrence and composition profile of 15 PFCs in surface water in the North Sea, Baltic Sea and Norwegian Sea.32 The composition profile in this area was influenced from local sources caused by human activities, whereas atmospheric deposition was negligible. Decreasing PFC concentrations with increasing distance from the coast was also found in surface water in the German Bight, which indicates that the rivers and coastal areas are the dominant sources of PFCs in the marine environment.93
The global occurrence of PFCs in open-ocean water was first described by Yamashita et al. (2005).5 Seawater samples were collected from the North and Mid Atlantic Ocean from 2002–2004. The concentration levels ranged from several tens pg per litre for perfluorohexane sulfonate (PFHxS), PFOS and perfluorononanoate (PFNA) to a few hundred pg per litre for PFOA. A similar study was carried out from 53° N to 30° S in the Atlantic Ocean in 2005.94 The concentration of PFOA and PFOS were in a range of a few tens pg per litre with a maximum concentration of 170 pg L−1 for PFOS. Overall, the ocean currents have a high influence on the occurrence of PFCs in the Atlantic Ocean.21 Concentrations of PFOS and PFOA reported in the Mid to South Pacific Ocean and the Indian Ocean were about one magnitude lower than in the North Atlantic Ocean.21,95 Furthermore, vertical profiles of several PFCs were studied in the Labrador Sea, Mid Atlantic Ocean, South Pacific Ocean and Japan Sea.22 It was hypothesised that PFCs could be transported globally with the thermohaline circulation system, and the open-ocean water is acting as a final sink for PFOS and PFOA.
In general, the concentration of PFOA is usually higher than of PFOS, which suggest that similar sources come from the urbanised/industrial coastal area. In addition, the higher level of PFOA in seawater could be explained by its higher water solubility,16 lower bioaccumulation potential10 and lower sorption potential to sediment.96
Fate and transport mechanisms
The environmental fate of PFCs results from the interplay of numerous processes including physical transport and multimedia partitioning. Different pathways of PFCs in the environment are possible. Volatile PFCs can be transported by the atmosphere, while ionic PFCs can enter the aquatic environment directly. From the product manufacturing processes, supply chains, product use, and disposal, PFCs can be released into the aquatic environment. Sources are thought to be dry and wet deposition, industrial and domestic WWTPs, landfill sites, and runoff from contaminated sites (see section ‘Sources and origins’).51–54,65,67 The proposed pathway of PFCs from the production and usage to the aqueous environment and biota is shown in Fig. 3.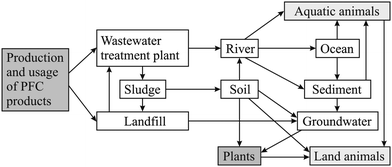 | ||
| Fig. 3 Environmental fate of PFCs in the aqueous environment. Note: the atmospheric pathway is not shown. | ||
The environmental fate is influenced by their physicochemical properties which vary depending on their chain length and functional groups. Ionic PFCs, like PFCAs and PFSAs, are very persistent, because of the strong bonding between the carbon and fluorine atom (>450 kJ mol−1) and the shielding of the carbon by the fluorine atoms.1 They have high water solubility, low pKa values and therefore dissociate at environmental relevant pH values. Because of the low vapour pressure of the ions, they will be primarily found in water or bound to particles, sediment and soil or bioaccumulate in the food web.1,10,17,96
Neutral PFCs are less persistent than the PFSAs and PFCAs and can be transformed by hydrolysis, photolysis and biodegradation.3,4 In addition, the neutral PFCs have a higher vapour pressure and lower water solubility in comparison to the PFSAs and PFCAs, which makes a long-range atmospheric transport for volatile neutral PFCs possible.2,3 Smog chamber experiments have shown that FTOHs can degrade by OH-initiated oxidation pathways, with the intermediates FTCAs and FTUCAs, to PFCAs in the atmosphere,2 and a lifetime of approximately 20 days for the FTOHs was estimated.97 Furthermore, laboratory studies showed that FASEs and FASAs can degrade to PFSAs and PFCAs in the atmosphere.3,61
The determination of partition coefficients and also the adsorption behaviour are vital to verify the environmental fate of PFCs. The adsorption behaviour of PFCs has been investigated in terms of the solid/water partition coefficient (Kd).96,98,99 The partitioning of PFCs in solid/water was determined in marine sediment cores and seawater in Tokyo Bay, Japan.96,98 In both studies, the perfluorocarbon chain length and functional group were the dominating parameter influencing the partitioning of PFCs. Hence, the short chain PFCAs (C < 7) were exclusively found in the dissolved phase, while long chain PFCAs (C ≥ 7), PFSAs, ethylperfluorooctane sulfonamidoacetic acid (EtFOSAA), and PFOSA appeared to bind more strongly to particles. In addition, the partitioning varied depending on the conditions (e.g., organic carbon content, pH value, metal ions), for example, an increasing sorption was found with increasing organic carbon content.
As a consequence, the short chain PFCs have a higher potential for aqueous long-range transport and PFSAs, EtFOSAA and PFOSA are distributed in biota or matrices in the abiotic environment like sediment, which could act as a sink for PFCs. The physicochemical characteristics also have an influence on the mechanism of long-range transport in the aqueous environment (e.g., sea-spray, microlayer, surface water, deep ocean water). However, the dominant transport pathways of individual PFCs to remote regions has not been conclusively characterised to date.
Current challenges in fate and global geochemical cycle processes
The complexity of the geochemical cycle processes has been omitted from many POP studies. Ideally, the knowledge of the source emission rates with the quantification of environmental reservoirs and final sink fluxes of PFCs can be combined to a global mass balance. However, it is a challenge to get accurate emission rates to improve our understanding of the global distribution of PFCs. Important point sources are WWTP effluents, from which PFCs are further transported by the rivers into the marine environment. As expected, there is a general gradient, with decreasing concentrations from WWTP effluents to the open-ocean water (Fig. 4).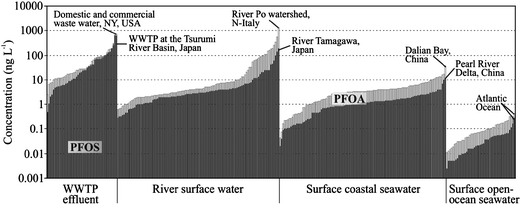 | ||
| Fig. 4 PFOA (white bars) and PFOS (black bars) concentrations in WWTP effluent,34,55–57,61,63,71,74 river surface water,56–58,64,87,103 surface coastal seawater23,95–97 and surface open-ocean seawater.5,21,93,94. | ||
The PFOA and PFOS concentrations were in the range of tens to hundreds of ng per litre in WWTP effluents.34,57,58WWTP-derived total fluxes in Japan were estimated to be 3.6, 2.6, 5.6, and 2.6 t year−1 for PFOS, perfluoroheptanoate (PFHpA), PFOA, and PFNA, respectively.59 An order of magnitude lower concentrations were found in river surface water, while the maximum concentration levels were in a similar range as in the WWTP effluents (see Fig. 4). Rivers are important input pathways of PFCs into the marine environment.79 The total flux of PFOA from the European rivers was estimated to be 14 t year−1, while the flux from the Yangtze River, China, was estimated to be 3.8 t year−1 for PFOA and 0.5 t year−1 for PFOS.79,92,100 Coastal areas are highly relevant in terms of PFC cycling since they are highly populated, however, the role of the coastal sediment which can act as a permanent sink or a source by resuspension is not fully understood for PFCs.96 The PFOA and PFOS concentrations ranged generally between hundreds of pg per litre to a few ng per litre in the coastal seawater.93,101 PFCs can be further transported in which the continental shelf is an interface between open-oceans and continents. The surface oceans were reported to be the most important reservoir for PFCs, however, the spatial distribution of PFCs in open-oceans seawater depends on the distance to sources (e.g., coastal area, rivers) and influence of the ocean currents.5,21 The highest concentrations of PFOA and PFOS were found in the North and Mid Atlantic Ocean and the Western Pacific Ocean, while the lowest concentrations were observed in the Central and Southern Pacific, Indian Ocean and the Southern Ocean.5,21,95,102 Interestingly, the ratio between PFOA/PFOS increased from 2.65 (WWTP effluents) to 3.10 (surface open-ocean seawater) indicating removal of PFOS during the transport process. The most likely removal process is the sedimentation because of the higher organic carbon normalized partition coefficient (log KOC) of PFOS (3.8 ± 0.1 cm3 g−1) in comparison to PFOA (1.9 ± 0.1 cm3 g−1).98
PFCs are very persistent, making them ideal tracers for ocean currents and globally transport by the thermohaline circulation system.22 Ocean currents and related dilution effects have a crucial influence on PFC distribution in seawater, in which industrial and coastal areas are considered a source of PFCs, and ocean waters are important sinks and for transportation of these compounds.21 Removal rates for PFOA from the surface water was estimated to be 0.2–33 t year−1.17 Vertical profiles of PFCs showed non-detectable or negligible concentrations in deep ocean water from Mid Atlantic Ocean, Japan Sea and South Pacific Ocean, which indicate very slow transportation by the ocean currents.22 In comparison to deep ocean water currents, the wind-driven and sea spray-mediated surface water transport is much faster.23
Several modelling and inventory studies have attempted to resolve the debate about the dominant transport pathway of PFCs to remote regions.15,17,46,103 In general, medium chain length PFCAs (C8 and C9) were transported dominantly via the ocean currents to the Arctic, while the atmospheric transport and subsequent degradation of FTOHs is the dominant source for the longer chain PFCAs (C ≥ 10).15 For the global inventory of individual PFCs, little data is available. The total inventory is estimated to be 110–10000 t for PFOA and 235–1770 t for PFOS in ocean seawater.17,46 However, PFOA can be emitted to the atmosphere by aerosol-mediated transport, which indicates long-range transport of PFOAvia the gas phase.16
Isomeric profiles of PFCs in seawater may help to distinguish between current and historical releases. Thus, the presence of branched isomers may indicate exposure from historical releases of ECF manufacturing processes42 or be a sign of local ECF production while the presence of exclusively linear isomers indicate telomerisation production processes. The observed isomer profile is consistent with the hypothesis that historical ECF-derived emissions are the major source of PFOA to the oceans,17 however, branched isomers could also be preferentially enriched in surface waters which may influence the observed pattern.
Based on temporal trend trends in polar bears (Ursus maritimus) and ringed seals, which showed increasing PFC concentrations in polar bears from East Greenland and decreasing concentrations in ringed seals from the Canadian Arctic, North America and Europe appear to be the major source regions for the Arctic region.70,104 The increasing PFOS concentrations in polar bears indicated that the ocean currents are the major source for the contamination, while the rapid decreasing concentrations in ringed seals were explained by the change of atmospheric deposition. Armitage et al. (2006) predicted that the PFOA emissions from point sources would decrease after 2005, but in the Arctic Ocean the PFOA concentration would increase further until approximately 2030 and then gradually decline.103 The time lag in the Arctic Ocean is caused by the long transport route to the Arctic. However, the temporal trend studies are only an indirect proof of the dominant pathway of PFCs and many uncertainties are still present in global transport models due to a lack of knowledge of the fate and global geochemical cycle processes of PFCs.
Conclusions and future research needs
This paper is a comprehensive, critical review of the occurrence and fate of PFCs in the aquatic environment. For the measurements a good quality assurance and control is necessary to make the data more accurate, reliable and comparable. Point sources (e.g., WWTP effluents) and diffuse sources (e.g., surface runoff) were identified. PFCs are further transported mainly by the rivers into the marine environment.79 Individual PFCs from various classes have been found in the ng per litre range in coastal seawater and in the pg per litre range in open-ocean water depending on the location and the compound.21,101 The surface ocean and atmosphere are reservoirs, while the deep ocean, deep soil and sediment are potential sinks of PFCs. However, the physical transport and multimedia partitioning depends on their physicochemical properties which vary depending on the chain length and functional group. In addition, the emissions of PFCs back into the biogeochemical cycling (e.g., resuspension from the sediment) is not fully understood. Several modelling studies have attempted to resolve the debate about the dominant transport pathway of PFCs to remote regions (i.e., atmospheric transport of precursors versus direct transportvia ocean currents) where the ocean currents have been identified as the dominant transport pathway.103 However, the models are often limited by the lack of emission information and the current models over simplify the oceanic vertical structure and transport which can bias flux estimations and response times. Recently, the quantification of branched isomers has become more important because of their toxic relevance40,41 and the PFC isomer pattern can be used to identify the dominant source as either historical or current releases of fluoropolymer products.42 However, the major contribution of organofluorine compounds was found to be unknown (60–90%), which indicate the presence of other PFCs in addition to the known PFCs.39Several areas of future research are needed before the environmental fate and behaviour of PFCs is fully understood. These include research on key loss processes and deposition, the relationship between sources and aqueous environment concentrations, solid/water partitioning or air–water exchange, transport mechanism and the extent to which PFCs undergo long-range global transport. In addition, appropriate time series measurements are necessary to investigate seasonality and long-term changes (e.g., changes from the C8- to C4-PFCs as the dominant compounds). These include the establishment of a global monitoring program for PFCs in river water and seawater (like the Global Atmospheric Passive Sampling (GAPS) study for air105) to get a better understanding of the relationship between sources, water concentration and the mechanism of long-range global transport.
References
-
E. Kissa, Fluorinated Surfactants and Repellents, Marcel Dekker, New York, 2001 Search PubMed
.
- D. A. Ellis, J. W. Martin, S. A. Mabury, A. O. De Silva, M. D. Hurley, M. D. Sulbaek Anderson and T. J. Wallington, Degradation of fluorotelomer alcohols: a likely atmospheric source of perfluorinated carboxylic acids, Environ. Sci. Technol., 2004, 38, 3316–3321 CrossRef CAS
- J. W. Martin, D. A. Ellis, S. A. Mabury, M. D. Hurley and T. J. Wallington, Atmospheric chemistry of perfluoroalkanesulfonamides: kinetic and product studies of the OH radical and Cl atom initiated oxidation of n-ethyl perfluorobutanesulfonamide, Environ. Sci. Technol., 2006, 40, 864–872 CrossRef CAS
- K. R. Rhoads, E. M.-L. Janssen, R. G. Luthy and C. S. Criddle, Aerobic biotransformation and fate of n-ethyl perfluorooctane sulfonamidoethanol (n-EtFOSE) in activated sludge, Environ. Sci. Technol., 2008, 42, 2873–2878 CrossRef CAS
- N. Yamashita, K. Kannan, S. Taniyasu, Y. Horii, G. Petrick and T. Gamo, A global survey of perfluorinated acids in oceans, Mar. Pollut. Bull., 2005, 51, 658–668 CrossRef CAS
- A. Jahnke, U. Berger, R. Ebinghaus and C. Temme, Latitudinal gradient of airborne polyfluorinated alkyl substances in the marine atmosphere between Germany and South Africa (53° N–33° S), Environ. Sci. Technol., 2007, 41, 3055–3061 CrossRef CAS
- S. K. Ostertag, B. A. Tague, M. M. Humphries, S. A. Tittlemier and H. M. Chan, Estimated dietary exposure to fluorinated compounds from traditional foods among Inuit in Nunavut, Canada, Chemosphere, 2009, 75, 1165–1172 CrossRef CAS
- J. P. Giesy and K. Kannan, Global distribution of perfluorooctane sulfonate in wildlife, Environ. Sci. Technol., 2001, 35, 1339–1342 CrossRef CAS
- K. Kannan, S. Corsolini, J. Falandysz, G. Fillmann, K. S. Kumar, B. G. Loganathan, M. Ali Mohd, J. Olivero, N. Van Wouwe, J. Ho Yang and K. M. Aldous, Perfluorooctanesulfonate and related fluorochemicals in human blood from several countries, Environ. Sci. Technol., 2004, 38, 4489–4495 CrossRef CAS
- J. W. Martin, S. A. Mabury, K. R. Solomon and D. C. G. Muir, Bioconcentration and tissue distribution of perfluorinated acids in rainbow trout (Oncorhynchus mykiss), Environ. Toxicol. Chem., 2003, 22, 196–204 CrossRef CAS
- S. Shaw, M. L. Berger, D. Brenner, L. Tao, Q. Wu and K. Kannan, Specific accumulation of perfluorochemicals in harbor seals (Phoca vitulina concolor) from the northwest Atlantic, Chemosphere, 2009, 74, 1037–1043 CrossRef CAS
- C. Lau, K. Anitole, C. Hodes, D. Lai, A. Pfahles-Hutchens and J. Seed, Perfluoroalkyl acids: a review of monitoring and toxicological findings, Toxicol. Sci., 2007, 99, 366–394 CrossRef CAS
- United Nations Environment Programme, The Environment in the News, 11 May 2009, from http://www.unep.org/cpi/briefs/(12/01/2010).
- U. Schenker, M. Scheringer, M. MacLeod, J. W. Martin, I. T. Cousins and K. Hungerbühler, Contribution of volatile precursor substances to the flux of perfluorooctanoate to the Arctic, Environ. Sci. Technol., 2008, 42, 3710–3716 CrossRef CAS
- J. M. Armitage, M. Macleod and I. T. Cousins, Comparative assessment of the global fate and transport pathways of long-chain perfluorocarboxylic acids (PFCAs) and perfluorocarboxylates (PFCs) emitted from direct sources, Environ. Sci. Technol., 2009, 43, 5830–5836 CrossRef CAS
- C. J. McMurdo, D. A. Ellis, E. Webster, J. Butler, R. D. Christensen and L. K. Reid, Aerosol enrichment of the surfactant PFO and mediation of the water–air transport of gaseous PFOA, Environ. Sci. Technol., 2008, 42, 3969–3974 CrossRef CAS
- K. Prevedouros, I. T. Cousins, R. C. Buck and S. H. Korzeniowski, Sources, fate and transport of perfluorocarboxylates, Environ. Sci. Technol., 2006, 40, 32–44 CAS
- S. P. J. Van Leeuwen and J. De Boer, Extraction and clean-up strategies for the analysis of poly- and perfluoroalkyl substances in environmental and human matrices, J. Chromatogr., A, 2007, 1153, 172–185 CrossRef CAS
- P. De Voogt and M. Sáez, Analytical chemistry of perfluoroalkylated substances, TrAC, Trends Anal. Chem., 2006, 25, 326–342 CrossRef CAS
- A. Jahnke and U. Berger, Trace analysis of per- and polyfluorinated alkyl substances in various matrices-How do current methods perform?, J. Chromatogr., A, 2009, 1216, 410–421 CrossRef CAS
- L. Ahrens, J. L. Barber, Z. Xie and R. Ebinghaus, Longitudinal and latitudinal distribution of perfluoroalkyl compounds in the surface water of the Atlantic Ocean, Environ. Sci. Technol., 2009, 43, 3122–3127 CrossRef CAS
- N. Yamashita, S. Taniyasu, G. Petrick, S. Wei, T. Gamo, P. K. S. Lam and K. Kannan, Perfluorinated acids as novel chemical tracers of global circulation of ocean waters, Chemosphere, 2008, 70, 1247–1255 CrossRef CAS
- X. Ju, Y. Jin, K. Sasaki and N. Saito, Perfluorinated surfactants in surface, subsurface water and microlayer from Dalian coastal waters in China, Environ. Sci. Technol., 2008, 42, 3538–3542 CrossRef CAS
- J. W. Martin, U. Berger, P. De Voogt, J. A. Field, J. Franklin, J. P. Giesy, T. Harner, D. C. G. Muir, B. Scott, M. Kaiser, U. Järnberg, K. C. Jones, S. A. Mabury, H. Schroeder, M. Simcik, C. Sottani, B. van Bavel, A. Kärrman, G. Lindstrom and S. Van Leeuwen, Analytical challenges hamper perfluoroalkyl research, Environ. Sci. Technol., 2004, 38, 248A–255A CrossRef CAS
- S. Taniyasu, K. Kannan, M. K. So, A. Gulkowska, E. Sinclair, T. Okazawa and N. Yamashita, Analysis of fluorotelomer alcohols, fluorotelomer acids, and short- and long-chain perfluorinated acids in water and biota, J. Chromatogr., A, 2005, 1093, 89–97 CrossRef CAS
- L. Ahrens, M. Plassmann, Z. Xie and R. Ebinghaus, Determination of polyfluoroalkyl compounds in water and suspended particulate matter in the River Elbe and North Sea, Germany, Front. Environ. Sci. Eng. China, 2009, 3, 152–170 Search PubMed
- H. P. H. Arp and K.-U. Goss, Gas/particle partitioning behavior of perfluorocarboxylic acids with terrestrial aerosols, Environ. Sci. Technol., 2009, 43, 8542–8547 CrossRef CAS
- C. A. Moody and J. A. Field, Determination of perfluorocarboxylates in groundwater impacted by fire-fighting activity, Environ. Sci. Technol., 1999, 33, 2800–2806 CrossRef CAS
- S. Taniyasu, K. Kannan, L. W. Y. Yeung, K. Y. Kwok, P. K. S. Lam and N. Yamashita, Analysis of trifluoroacetic acid and other short-chain perfluorinated acids (C2–C4) in precipitation by liquid chromatography–tandem mass spectrometry: Comparison to patterns of long-chain perfluorinated acids (C5–C18), Anal. Chim. Acta, 2008, 619, 221–230 CrossRef CAS
-
Iso, Guide to the Expression of Uncertainty in Measurement, International Organization for Standardization, Geneva, Switzerland, 1995, ISBN 92-67-10188-9 Search PubMed
- C. González-Barreiro, E. Martínez-Carballo, A. Sitka, S. Scharf and O. Gans, Method optimization for determination of selected perfluorinated alkylated substances in water samples, Anal. Bioanal. Chem., 2006, 386, 2123–2132 CrossRef CAS
- L. Ahrens, W. Gerwinski, N. Theobald and R. Ebinghaus, Sources of polyfluoroalkyl compounds in the North Sea, Baltic Sea and Norwegian Sea: Evidence from their spatial distribution in surface water, Mar. Pollut. Bull., 2010, 60, 255–260 CrossRef CAS
- C. R. Powley, S. W. George, T. W. Ryan and R. C. Buck, Matrix effect-free analytical methods for determination of perfluorinated carboxylic acids in environmental matrixes, Anal. Chem., 2005, 77, 6353–6358 CrossRef CAS
- M. M. Schultz, D. F. Barofsky and J. A. Field, Quantitative determination of fluorinated alkyl substances by large-volume-injection liquid chromatography tandem mass spectrometry-characterization of municipal wastewaters, Environ. Sci. Technol., 2006, 40, 289–295 CAS
- U. Berger, I. Langlois, M. Oehme and R. Kallenborn, Comparison of three types of mass spectrometer for high-performance liquid chromatography/mass spectrometry analysis of perfluoroalkylated substances and fluorotelomer alcohols, Eur. J. Mass Spectrom., 2004, 10, 579–588 CrossRef CAS
- N. Yamashita, K. Kannan, S. Taniyasu, Y. Horii, T. Okazawa, G. Petrick and T. Gamo, Analysis of perfluorinated acids at parts-per-quadrillion levels in seawater using liquid chromatography-tandem mass spectrometry, Environ. Sci. Technol., 2004, 38, 5522–5528 CrossRef CAS
- G. Arsenault, B. Chittim, A. McAlees, R. McCrindle, N. Riddell and B. Ye, Some issues relating to the use of perfluorooctanesulfonate (PFOS) samples as reference standards, Chemosphere, 2008, 70, 616–625 CrossRef CAS
- S. P. J. Van Leeuwen, C. P. Swart and J. De Boer, Significant improvements in the analysis of perfluorinated compounds in water and fish: results from an interlaboratory method evaluation study, J. Chromatogr., A, 2009, 1216, 401–409 CrossRef CAS
- Y. Miyake, N. Yamashita, P. Rostkowski, M. K. So, S. Taniyasu, P. K. S. Lam and K. Kannan, Determination of trace levels of total fluorine in water using combustion ion chromatography for fluorine: a mass balance approach to determine individual perfluorinated chemicals in water, J. Chromatogr., A, 2007, 1143, 98–104 CrossRef CAS
- J. P. Benskin, A. O. De Silva, L. J. Martin, G. Arsenault, R. McCrindle, N. Riddell, S. A. Mabury and J. W. Martin, Disposition of perfluorinated acid isomers in sprague-dawley rats: part 1: single dose, Environ. Toxicol. Chem., 2009, 28, 542–554 CrossRef CAS
- A. O. De Silva, J. P. Benskin, L. J. Martin, G. Arsenault, R. McCrindle, N. Riddell, S. A. Mabury and J. W. Martin, Disposition of perfluorinated acid isomers in sprague-dawley rats: part 2: subchronic dose, Environ. Toxicol. Chem., 2009, 28, 555–567 CrossRef CAS
- A. O. De Silva and S. A. Mabury, Isomer distribution of perfluorocarboxylates in human blood: potential correlation to source, Environ. Sci. Technol., 2006, 40, 2903–2909 CrossRef CAS
- S. P. J. Van Leeuwen, A. Kärrman, B. Van Bavel, J. De Boer and G. Lindström, Struggle for quality in determination of perfluorinated contaminants in environmental and human samples, Environ. Sci. Technol., 2006, 40, 7854–7860 CrossRef CAS
- L. Ahrens, K. Vorkamp, P. Lepom, P. Bersuder, N. Theobald, R. Ebinghaus, R. Bossi, J. L. Barber and E. McGovern, Determination of perfluoroalkyl compounds in water, sediment, and biota, ICES Techniques in Marine Environmental Sciences, 2010, 48, 1–17 Search PubMed
- H. Frank, E. H. Christoph, O. Holm-Hansen and J. L. Bullister, Trifluoroacetate in ocean waters, Environ. Sci. Technol., 2002, 36, 12–15 CrossRef CAS
- A. G. Paul, K. C. Jones and A. J. Sweetman, A first global production, emission, and environmental inventory for perfluorooctane sulfonate, Environ. Sci. Technol., 2009, 43, 386–392 CrossRef CAS
- M. M. Schultz, D. F. Barofsky and J. A. Field, Fluorinated alkyl surfactants, Environ. Eng. Sci., 2003, 20, 487–501 CrossRef CAS
- M. J. A. Dinglasan-Panlilio and S. A. Mabury, Significant residual fluorinated alcohols present in various fluorinated materials, Environ. Sci. Technol., 2006, 40, 1447–1453 CrossRef CAS
-
U.S. Environmental Protection Agency, PFOA Stewardship Program, 2006, Docket EPA-HQ-OPPT-2006-0621 Search PubMed
- European Parliament Council, European Community Directive 2006/122/ECOF, Off. J. Eur. Union, 2006, L 372, 32–34 Search PubMed
- S.-K. Kim and K. Kannan, Perfluorinated acids in air, rain, snow, surface runoff, and lakes: relative importance of pathways to contamination of urban lakes, Environ. Sci. Technol., 2007, 41, 8328–8334 CrossRef CAS
- M. Loewen, T. Halldorson, F. Wang and G. Tomy, Fluorotelomer carboxylic acids and PFOS in rainwater from an urban center in Canada, Environ. Sci. Technol., 2005, 39, 2944–2951 CrossRef CAS
- B. Boulanger, A. M. Peck, J. L. Schnoor and K. C. Hornbuckle, Mass budget of perfluorooctane surfactants in Lake Ontario, Environ. Sci. Technol., 2005, 39, 74–79 CrossRef CAS
- N. L. Stock, V. I. Furdui, D. C. G. Muir and S. A. Mabury, Perfluoroalkyl contaminants in the Canadian Arctic: evidence of atmospheric transport and local contamination, Environ. Sci. Technol., 2007, 41, 3529–3536 CrossRef CAS
- M. M. Schultz, C. P. Higgins, C. A. Huset, R. G. Luthy, D. F. Barofsky and J. A. Field, Fluorochemical mass flows in a municipal wastewater treatment facility, Environ. Sci. Technol., 2006, 40, 7350–7357 CrossRef CAS
- E. Sinclair and K. Kannan, Mass loading and fate of perfluoroalkyl surfactants in wastewater treatment plants, Environ. Sci. Technol., 2006, 40, 1408–1414 CrossRef CAS
- L. Ahrens, S. Felizeter, R. Sturm, Z. Xie and R. Ebinghaus, Polyfluorinated compounds in effluents of waste water treatment plants and surface water along the River Elbe, Germany, Mar. Pollut. Bull., 2009, 58, 1326–1333 CrossRef CAS
- C. A. Huset, A. C. Chiaia, D. F. Barfosky, N. Jonkers, H.-P. E. Kohler, C. Ort, W. Giger and J. A. Field, Occurrence and mass flows of fluorochemicals in the Glatt Valley watershed, Switzerland, Environ. Sci. Technol., 2008, 42, 6369–6377 CrossRef CAS
- M. Murakami, E. Imamura, H. Shinohara, K. Kiri, Y. Muramatsu, A. Harada and H. Takada, Occurence and sources of perfluorinated surfactants in rivers in Japan, Environ. Sci. Technol., 2008, 42, 6566–6572 CrossRef CAS
- B. G. Loganathan, K. S. Sajwan, E. Sinclair, K. S. Kumar and K. Kannan, Perfluoroalkyl sulfonates and perfluorocarboxylates in two wastewater treatment facilities in Kenntucky and Georgia, Water Res., 2007, 41, 4611–4620 CrossRef CAS
- J. C. D'eon, M. D. Hurley, T. J. Wallington and S. A. Mabury, Atmospheric chemistry of n-methyl perfluorobutane sulfonamidoethanol, C4F9SO2N(CH3)CH2CH2OH: Kinetics and mechanism of reaction with OH, Environ. Sci. Technol., 2006, 40, 1862–1868 CrossRef CAS
- A. M. Becker, S. Gerstmann and H. Frank, Perfluorooctane surfactants in waste waters, the major source of river pollution, Chemosphere, 2008, 72, 115–121 CrossRef CAS
- A. Y.-C. Lin, S. C. Panchangam and C.-C. Lo, The impact of semiconductor, electronics and optoelectronic industries on downstream perfluorinated chemical contamination in Taiwanese rivers, Environ. Pollut., 2009, 157, 1365–1372 CrossRef CAS
- Y. Zushi, T. Takeda and S. Masunaga, Existence of nonpoint source of perfluorinated compounds and their loads in the Tsurumi River basin, Japan, Chemosphere, 2008, 71, 1566–1573 CrossRef CAS
- A. Möller, L. Ahrens, R. Sturm and R. Ebinghaus, Identification of point sources of polyfluoroalkyl compounds (PFCs) along the River Rhine watershed and their transportation into the North Sea, Coastline Reports, 2009, 13, 143–154 Search PubMed
- R. Bossi, J. Strand, O. Sortkjaer and M. M. Larson, Perfluorinated compounds in Danish wastewater treatment plants and aquatic environments, Environ. Int., 2008, 34, 443–450 CrossRef CAS
- J. Busch, L. Ahrens, R. Sturm and R. Ebinghaus, Polyfluoroalkyl compounds in landfill leachates, Environ. Pollut., 2010, 158, 1467–1471 CrossRef CAS
- A. Jahnke, L. Ahrens, R. Ebinghaus and C. Temme, Urban versus remote air concentrations of fluorotelomer alcohols and other polyfluorinated alkyl substances in Germany, Environ. Sci. Technol., 2007, 41, 745–752 CrossRef CAS
- B. F. Scott, C. Spencer, S. A. Mabury and D. C. G. Muir, Poly and perfluorinated carboxylates in North American precipitation, Environ. Sci. Technol., 2006, 40, 7167–7174 CrossRef CAS
- C. Butt, D. C. Muir, I. Stirling, M. Kwan and S. A. Mabury, Rapid response of Arctic ringed seals to changes in perfluoroalkyl production, Environ. Sci. Technol., 2007, 41, 42–49 CrossRef CAS
- D. Skutlarek, M. Exner and H. Färber, Perfluorinated surfactants in surface and drinking waters, Environ. Sci. Pollut. Res., 2006, 13, 299–307 CrossRef CAS
- M. Murakami, H. Shinohara and H. Takada, Evaluation of wastewater and street runoff as sources of perfluorinated surfactants (PFSs), Chemosphere, 2009, 74, 487–493 CrossRef CAS
- C. A. Moody and J. A. Field, Perfluorinated surfactants and the environmental implications of their use in fire-fighting foams, Environ. Sci. Technol., 2000, 34, 3864–3870 CrossRef CAS
- C. A. Moody, J. W. Martin, W. C. Kwan, D. C. G. Muir and S. A. Mabury, Monitoring perfluorinated surfactants in biota and surface water samples following an accidental release of fire-fighting foam into Etobicoke Creek, Environ. Sci. Technol., 2002, 36, 545–551 CrossRef CAS
- M. H. Plumlee, J. Larabee and M. Reinhard, Perfluorochemicals in water reuse, Chemosphere, 2008, 72, 1541–1547 CrossRef CAS
- R. Loos, B. M. Gawlik, G. Locoro, E. Rimaviciute, S. Contini and G. Bidoglio, EU-wide survey of polar organic persistent pollutants in European river waters, Environ. Pollut., 2009, 157, 561–568 CrossRef CAS
- A. O. De Silva and S. A. Mabury, Isolating Isomers of perfluorocarboxylates in polar bears (Ursus maritimus) from two geographical locations, Environ. Sci. Technol., 2004, 38, 6538–6545 CrossRef CAS
- J. W. Martin, M. M. Smithwick, B. M. Braune, P. F. Hoekstra, D. C. G. Muir and S. A. Mabury, Identification of long-chain perfluorinated acids in biota from the Canadian Arctic, Environ. Sci. Technol., 2004, 38, 373–380 CrossRef CAS
- M. S. McLachlan, K. E. Holmstrom, M. Reth and U. Berger, Riverine discharge of perfluorinated carboxylates from the European continent, Environ. Sci. Technol., 2007, 41, 7260–7265 CrossRef CAS
- C. J. Young, V. I. Furdui, J. Franklin, R. M. Koerner, D. C. Muir and S. A. Mabury, Perfluorinated acids in arctic snow: new evidence for atmospheric formation, Environ. Sci. Technol., 2007, 41, 3455–3461 CrossRef CAS
- A. Römpp, O. Klemm, W. Fricke and H. Frank, Haloacetates in fog and rain, Environ. Sci. Technol., 2001, 35, 1294–1298 CrossRef CAS
- M. A. M. Mahmoud, A. Kärrman, S. Oono, K. H. Harada and A. Koizumi, Polyfluorinated telomers in precipitation and surface water in an urban area of Japan, Chemosphere, 2009, 74, 467–472 CrossRef CAS
- D. Trudel, L. Horowitz, M. Wormuth, M. Scheringer, I. T. Cousins and K. Hungerbuhler, Estimating consumer exposure to PFOS and PFOA, Risk Anal., 2008, 28, 251–269 Search PubMed
- K. Harada, N. Saito, K. Sasaki, K. Inoue and A. Koizumi, Perfluorooctane sulfonate contamination of drinking water in the Tama River, Japan: estimated effects on resident serum levels, Bull. Environ. Contam. Toxicol., 2003, 71, 31–36 CAS
- R. Loos, J. Wollgast, T. Huber and G. Hanke, Polar herbicides, pharmaceutical products, perfluorooctanesulfonate (PFOS), perfluorooctanoate (PFOA), and nonylphenol and its carboxylates and ethoxylates in surface and tap waters around Lake Maggiore in Northern Italy, Anal. Bioanal. Chem., 2007, 387, 1469–1478 CrossRef CAS
- C. A. Moody, W. C. Kwan, J. W. Martin, D. C. G. Muir and S. A. Mabury, Determination of perfluorinated surfactants in surface water samples by two independent analytical techniques: liquid chromatography/tandem mass spectrometry and 19F NMR., Anal. Chem., 2001, 73, 2200–2206 CrossRef CAS
- K. J. Hansen, H. O. Johnson, J. S. Eldridge, J. L. Butenhoff and L. A. Dick, Quantitative characterization of trace levels of PFOS and PFOA in the Tennessee River, Environ. Sci. Technol., 2002, 36, 1681–1685 CrossRef CAS
- S. Nakayama, L. Helfant, P. Egeghy, X. Ye and A. B. Lindstrom, Perfluorinated compounds in the cape fear drainage basin in North Carolina, Environ. Sci. Technol., 2007, 41, 5271–5276 CrossRef CAS
- B. Boulanger, J. Vargo, J. L. Schnoor and K. C. Hornbuckle, Detection of perfluorooctane surfactants in Great Lakes water, Environ. Sci. Technol., 2004, 38, 4064–4070 CrossRef CAS
- F. Orata, N. Quinete, F. Werres and R.-D. Wilken, Determination of Perfluorooctanoic Acid and Perfluorooctane Sulfonate in Lake Victoria Gulf Water, Bull. Environ. Contam. Toxicol., 2009, 82, 218–222 CrossRef CAS
- S. Taniyasu, K. Kannan, Y. Horii, N. Hanari and N. Yamashita, A survey of perfluorooctane sulfonate and related perfluorinated organic compounds in water, fish, birds, and humans from Japan, Environ. Sci. Technol., 2003, 37, 2634–2639 CrossRef CAS
- M. K. So, Y. Miyake, W. Y. Yeung, Y. M. Ho, S. Taniyasu, P. Rostkowski, N. Yamashita, B. S. Zhou, X. J. Shi, J. X. Wang, J. P. Giesy, H. Yu and P. K. S. Lam, Perfluorinated compounds in the Pearl River and Yangtze River of China, Chemosphere, 2007, 68, 2085–2095 CrossRef CAS
- L. Ahrens, S. Felizeter and R. Ebinghaus, Spatial distribution of polyfluoroalkyl compounds in seawater of the German Bight, Chemosphere, 2009, 76, 179–184 CrossRef CAS
-
N. Theobald, W. Gerwinski, A. Jahnke, Occurrence of perfluorinated organic acids in surface sea-water of the East Atlantic Ocean between 53° north and 30° south, 2007, Poster presentation at the SETAC Europe Annual Meeting 20–24 May 2007 in Porto, Portugal Search PubMed
- S. Wei, L. Q. Chen, S. Taniyasu, M. K. So, M. B. Murphy, N. Yamashita, L. W. Y. Yeung, P. K. S. Lam, S. Wei and L. Q. Chen, Distribution of perfluorinated compounds in surface seawaters between Asia and Antarctica, Mar. Pollut. Bull., 2007, 54, 1813–1838 CrossRef CAS
- L. Ahrens, N. Yamashita, L. W. Y. Yeung, S. Taniyasu, Y. Horii, P. K. S. Lam and R. Ebinghaus, Partitioning behaviour of per- and polyfluoroalkyl compounds between pore water and sediment in two sediment cores from Tokyo Bay, Japan, Environ. Sci. Technol., 2009, 43, 6969–6975 CrossRef CAS
- D. A. Ellis, J. W. Martin, S. A. Mabury, M. D. Hurley, M. D. Sulbaek Anderson and T. J. Wallington, Atmospheric lifetime of fluorotelomer alcohols, Environ. Sci. Technol., 2003, 37, 3816–3820 CrossRef CAS
- L. Ahrens, S. Taniyasu, L. W. Y. Yeung, N. Yamashita, P. K. S. Lam and R. Ebinghaus, Distribution of polyfluoroalkyl compounds in water, suspended particulate matter and sediment from Tokyo Bay, Japan, Chemosphere, 2010, 79, 266–272 CrossRef CAS
- C. P. Higgins and R. G. Luthy, Sorption of perfluorinated surfactants on sediment, Environ. Sci. Technol., 2006, 40, 7251–7256 CrossRef CAS
- L. W. Y. Yeung, N. Yamashita, S. Taniyasu, P. K. S. Lam, R. K. Sinha, D. V. Borole and K. Kannan, A survey of perfluorinated compounds in surface water and biota including dolphins from the Ganges River and in other waterbodies in India, Chemosphere, 2009, 76, 55–62 CrossRef CAS
- M. K. So, S. Taniyasu, N. Yamashita, J. P. Giesy, J. Zheng, Z. Fang, S. H. Im and P. K. S. Lam, Perfluorinated compounds in coastal waters of Hong Kong, South China, and Korea, Environ. Sci. Technol., 2004, 38, 4056–4063 CrossRef CAS
- L. Ahrens, Z. Xie and R. Ebinghaus, Distribution of perfluoroalkyl compounds in seawater from Northern Europe, Atlantic Ocean, and Southern Ocean, Chemosphere, 2010, 78, 1011–1016 CrossRef CAS
- J. Armitage, I. T. Cousins, R. C. Buck, K. Prevedouros, M. H. Russell, M. Macleod and S. H. Korzeniowski, Modeling global-scale fate and transport of perfluorooctanoate emitted from direct sources, Environ. Sci. Technol., 2006, 40, 6969–6975 CrossRef CAS
- R. Dietz, R. Bossi, F. F. Riget, C. Sonne and E. W. Born, Increasing perfluoroalkyl contaminants in East Greenland polar bears (Ursus maritimus): a new toxic threat to the Arctic bears, Environ. Sci. Technol., 2008, 42, 2701–2707 CrossRef CAS
- K. Pozo, T. Harner, F. Wania, D. C. G. Muir, K. C. Jones and L. A. Barrie, Toward a global network for persistent organic pollutants in air: results from the GAPS Study, Environ. Sci. Technol., 2006, 40, 4867–4873 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2011 |