Production of green aromatics and olefins by catalytic fast pyrolysis of wood sawdust
Torren R.
Carlson
,
Yu-Ting
Cheng
,
Jungho
Jae
and
George W.
Huber
*
Department of Chemical Engineering, University of Massachusetts, 159, Goessmann Laboratory, Amherst, MA 01003, USA. E-mail: huber@ecs.umass.edu; Tel: +1 4135450276
First published on 26th October 2010
Abstract
Catalytic fast pyrolysis of pine wood sawdust and furan (a model biomass compound) with ZSM-5 based catalysts was studied with three different reactors: a bench scale bubbling fluidized bed reactor, a fixed bed reactor and a semi-batch pyroprobe reactor. The highest aromatic yield from sawdust of 14% carbon in the fluidized bed reactor was obtained at low biomass weight hourly space velocities (less than 0.5 h−1) and high temperature (600 °C). Olefins (primarily ethylene and propylene) were also produced with a carbon yield of 5.4% carbon. The biomass weight hourly space velocity and the reactor temperature can be used to control both aromatic yield and selectivity. At low biomass WHSV the more valuable monocyclic aromatics are produced and the formation of less valuable polycyclic aromatics is inhibited. Lowering the reaction temperature also results in more valuable monocyclic aromatics. The olefins produced during the reaction can be recycled to the reactor to produce additional aromatics. Propylene is more reactive than ethylene. Co-feeding propylene to the reactor results in a higher aromatic yield in both continuous reactors and higher conversion of the intermediate furan in the fixed bed reactor. When olefins are recycled aromatic yields from wood of 20% carbon can be obtained. After ten reaction–regeneration cycles there were metal impurities deposited on the catalyst, however, the acid sites on the zeolite are not affected. Of the three reactors tested the batch pyroprobe reactor yielded the most aromatics, however, the aromatic product is largely naphthalene. The continuous reactors produce less naphthalene and the sum of aromatics plus olefin products is higher than the pyroprobe reactor.
Broader contextCatalytic fast pyrolysis (CFP) is a promising technology to directly convert solid biomass to gasoline-range aromatics that fit into the current infrastructure. CFP involves the rapid heating of biomass (∼500 °C s−1) in an inert atmosphere to intermediate temperatures (400 to 600 °C) in the presence of zeolite catalysts. During CFP, biomass is converted in a single step to produce gasoline-range aromatics which are compatible with the gasoline of the current market. CFP has many advantages over other conversion processes including short residence times (2–10 s) and inexpensive catalysts. In this study we show that CFP can be performed in a continuous fluidized bed reactor with real biomass feeds. We also show how process parameters can be used to control both the yield and selectivity for the aromatic products. |
1.0 Introduction
Catalytic fast pyrolysis (CFP), which involves the pyrolysis of biomass in the presence of a zeolite catalyst, is a promising technology for the direct conversion of solid biomass into gasoline range aromatics.1–7 There are significant advantages to this approach for biomass conversion including (1) all the desired chemistry occurs in one single reactor, (2) in-expensive silica–alumina catalyst are used, (3) no water is required for this process, (4) CFP can be used to process a range of different lignocellulosic feedstocks, (5) biomass pretreatment is simple (drying and grinding) and (6) the process occurs in fluidized bed reactors which are already used commercially today in almost every petroleum refinery. Furthermore CFP produces products (aromatics and olefins) that already fit into existing infrastructure. The petroleum industry uses six major petrochemicals as feedstocks. CFP can produce five of these six major petrochemicals including benzene, toluene, xylene, ethylene and propylene.The reaction pathways for the conversion of cellulose into aromatics by CFP are shown in Fig. 1. The first step is the thermal decomposition (or pyrolysis) of cellulose to anhydrosugars and other condensable oxygenated products like dihydroxyacetone and glyceraldehyde.8–12 The formation of these oxygenated pyrolysis products is a high activation energy process compared to the formation of coke, which also produces CO2 and H2O. When high heating rates and temperatures are used the anhydrosugars are the primary pyrolysis products.13,14 The anhydrosugars are relatively thermally stable and do not form large amounts of coke in the gas phase.15 However, the anhydrosugars can undergo dehydration and re-arrangement reactions to form furans, smaller aldehydes and H2O. These reactions can happen in either the gas phase or in the presence of a catalyst.13 These intermediate oxygenates then diffuse into the zeolite catalyst pores and through a series of decarbonylation, decarboxylation, dehydration, and oligomerization reactions form both monocyclic aromatics and olefins.13,16,17 The major competing reaction with the formation of aromatics is the formation of coke from the polymerization of the furans. The aromatic formation reaction proceeds through a common intermediate or “hydrocarbon pool” within the zeolite framework.18–22 The polycyclic aromatics such as naphthalene are formed in a second series reaction where the monocyclic aromatics further react with another oxygenate.18
 | ||
| Fig. 1 Overall reaction chemistry for the CFP of cellulose. | ||
We have previously studied the chemistry of CFP using glucose as a model compound for the cellulose portion of biomass.13,18 We have shown that under optimized conditions in a pyroprobe microreactor 32% of the carbon in the glucose can be converted into aromatics. The yield and selectivity for aromatics from the CFP of glucose show the potential for the CFP process,13 however, the micropyroprobe reactor cannot economically be scaled up into a larger reactor. However, as we will show in this paper CFP can be done efficiently in a fluidized bed reactor. Fluidized bed reactors have been proven in a vast number of processes across industry due to their excellent mass and heat transfer properties, scalability and simplicity of operation.23–26
Several researchers have also performed catalytic pyrolysis in continuous fluidized bed reactors.1–3,27–29 In early studies aromatics were produced by first pyrolyzing wood in a non-catalytic fluidized bed reactor followed by a secondary fixed bed catalytic reactor to convert the primary pyrolysis vapors.27–29 In general the two reactor approaches yielded more coke and less aromatics when compared with the later single stage studies. Of the single stage studies Olazar and coworkers2 reported aromatic yields of 12% carbon in a conical spouted bed reactor using ZSM-5 catalyst. In this reactor setup the bottom section of the reactor is conical in shape and a high velocity stream of gas (the spout) induces circulation within the catalyst bed. Aho et al.1 tested several types of zeolites for the catalytic pyrolysis of softwood in a cylindrical bubbling fluidized bed reactor. They reported that β-zeolite, mordenite, Y-zeolite, and ZSM-5 all produced different product spectra in the resulting bio-oil. The addition of ZSM-5 significantly decreased the amount of acids and alcohols in the bio-oil while the amount of ketones increased. Lappas and collaborators3 reported on the use of a lab scale FCC riser reactor for the catalytic pyrolysis of pine wood with a commercial fluid catalytic cracking catalyst and a commercial ZSM-5 additive. They reported that addition of catalyst increased the yield of water, non-condensable gases and char. The bio-oil obtained was of lower oxygen content and therefore they proposed of better quality.
The objective of this paper is to study CFP of pine wood in a bubbling fluidized bed reactor and compare these results with CFP in a fixed bed reactor and a pyroprobe reactor. The effects of temperature, biomass weight hourly space velocity (WHSV) and reaction time on the product yield and selectivity are studied. In addition we test the catalytic properties before and after reaction to determine if impurities in the biomass poison catalytic sites. Biomass contains minerals that may poison zeolite catalysts during CFP.30 Several researchers have studied the effect of salts on the primary pyrolysis reactions31 and metal catalyzed hydrogenation reactions,32 however, none of the previous researchers studied the effect of the minerals on zeolite catalyst stability. In the fixed bed reactor we study furan conversion over ZSM-5 and compare these results to CFP in a fluidized bed reactor. We study the effects of co-feeding olefins in both the fluidized bed and fixed bed reactors. The fluidized bed and fixed bed results are also compared to a pyroprobe microreactor to show how CFP can change with different reactor types and catalysts.
2.0 Experimental
2.1 Feed
Furan (Sigma-Aldrich) was used as feedstock without any pretreatment. The wood for the pyroprobe and fluidized bed studies was eastern pine sawdust. The elemental analysis of the wood feed is shown in Table 1. The chemical formula for the untreated wood used is therefore approximately C4H6O3. The moisture content of the feed was determined to be 4 wt% by the mass difference between a 5 g sample of feed before and after drying in a 95 °C oven overnight. On a dry basis the approximate chemical formula of the wood is C3.8H5.8O2.7. The ash content was determined by the mass difference between a 1 g sample of feed before and after burning in air at 600 °C for 5 hours in a muffle oven.| Elemental analysis/wt% | |||
|---|---|---|---|
| C | H | Oa | Ash |
| a By balance. | |||
| 46.19 | 6.02 | 47.29 | 0.47 |
2.2 Catalyst
For the fixed bed studies ZSM-5 powder in the proton form (Zeolyst CBV 3024E, SiO2/Ai2O3 = 30) was used. The as-received ZSM-5 catalyst was sieved to 425–800 µm before reaction. Prior to reaction the catalyst was calcined for 5 hours in the fixed bed reactor at 600 °C with flowing air (Airgas) at 60 sccm. The catalyst used in the fluidized bed experiments was a commercial spray dried ZSM-5 catalyst (BioCat1, Grace Davison). The catalyst was calcined in the reactor for 4 hours at 600 °C in 1200 sccm flowing air prior to reaction. For reactions in the pyroprobe batch reactor both catalysts were calcined in a muffle oven at 600 °C for 5 hours. The Grace Davison catalyst was characterized before and after ten reaction/regeneration cycles by XRD (PANalytical X'Pert Pro Material Research Diffractometer) and SEM (JEOL JSM-5400 Scanning electron microscope). Ammonia TPD curves of the fresh and spent catalyst were measured using a ChemBET Pulsar TPR/TPD system (Quantachrome Instruments). The samples were degassed at 450 °C in 60 sccm of flowing helium. Ammonia was adsorbed at 100 °C for 20 minutes followed by purging with helium for one hour. TPD was performed from 100 to 600 °C with a temperature ramp rate of 10 °C min−1. FTIR spectra of adsorbed ammonia at 150 °C were taken using a DRIFTS cell (Harrick Scientific) with an Equinox 55 FTIR spectrometer (Bruker).2.3 Pyroprobe
Fast pyrolysis experiments were conducted using a model 2000 pyroprobe analytical pyrolizer (CDS Analytical Inc.). The probe is a computer controlled resistively heated element which holds an open ended quartz tube (pictured in Fig. 2). Powdered samples are held in the tube with loose quartz wool packing; during pyrolysis vapors flow from the open ends of the quartz tube into a larger cavity (the pyrolysis interface) with a helium carrier gas stream. | ||
| Fig. 2 Diagram of the pyroprobe reactor setup. On the left a schematic cross-section of the prepared sample (not to scale). Powdered reactants and catalysts are held with loose quartz wool packing. Pictured on the right is the resistively heated element which holds the sample tube (2 mm × 25 mm). During reaction product vapors flow from the open ends of the sample tube into the GC/MS interface via a helium sweeper gas stream. | ||
The carrier gas stream is routed to a model 5890 gas chromatograph (GC) interfaced with a Hewlett Packard model 5972A mass spectrometer (MS). The pyrolysis interface was held at 100 °C and the GC injector temperature used was 275 °C. Helium was used as the inert pyrolysis gas as well as the carrier gas for the GC/MS system.
Powdered reactants were prepared by physically mixing feed and catalyst. For a typical run 8–15 mg of reactant–catalyst mixture were used. Both the solid feed and the catalyst were sifted to <140 mesh before mixing. The physical mixtures of wood and ZSM-5 were prepared with a catalyst to wood weight ratio of 19. The mixture of furan and ZSM-5 was prepared with catalyst to furan weight ratio of 19.
2.4 Fixed bed
The fixed bed reactor was built from a ½ inch diameter quartz tube. Sieved ZSM-5 powders were held in the reactor by quartz beads (250–425 µm) and quartz wool. Typically ∼26 mg of catalyst are used in the catalyst bed. The reactor temperature was measured using a thermocouple inserted on top of the catalyst bed. Prior to reactions, the catalyst bed was calcined as described above. After calcination the reactor was flushed by helium (Airgas, ultra-high purity) for 10 min. The helium then was switched to bypass the reactor, and the inlet and outlet valves of the reactor were closed. Furan was pumped into the helium stream by a syringe pump (Fisher, KDS100) at a rate of 0.29 mL h−1. The carrier gas was controlled at 204 sccm yielding a furan partial pressure 6 Torr. Prior to the run the furan bypassed the reactor for 30 min before switching the helium stream to go through the reactor. An ice-water bath condenser was used to trap the heavy products. Gas phase products were collected by air bags. All runs were done at atmospheric pressure. After reaction, the reactor was flushed by helium with the flow rate of 30 sccm for 20 min at the reaction temperature. Again, the effluent was collected by an air bag, and the heavy hydrocarbons were condensed in the condenser.After flushing the reactor was cooled to room temperature with 10 sccm helium flow. Condensed products were extracted by 10 mL ethanol from the condensers to obtain the liquid products. Both liquid and gas products were identified by GC-MS (Shimadzu-2010) and quantified by GC-FID (Shimadzu 2014 for gas samples and HP-7890 for liquid samples). The spent catalyst was removed from the reactor and was subjected to TGA analysis to obtain coke amount. In our study, less than 0.05% carbon or the products were collected in the liquid trap. The majority of the products were in either the gas phase or coke deposited on the catalyst.
2.5 Fluidized bed reactor
A schematic of the fluidized bed reactor system is shown in Fig. 3. The fluidized bed reactor is a 2 in 316 stainless steel tube 10 in tall. The wood feed was injected by a stainless steel auger into the side of the reactor from a sealed feed hopper. The auger is turned by a variable speed motor to provide a constant feed flow rate during reaction. The feed system was calibrated for different flow rates before reaction using a balance and a stopwatch. Then to maintain an inert environment in the reactor the hopper is swept with helium at a rate of 200 sccm. The wood used was sieved to a particle size of 0.25–1 mm before loading it into the hopper. The catalyst bed is supported by a distributor plate made from stacked 316 stainless steel mesh (300 mesh). During the reaction, catalyst is fluidized via a helium gas stream controlled by a mass flow controller to a flow rate of 1200 sccm. The gas flow rate and catalyst used resulted in a bubbling fluidized bed flow regime.23 Both the reactor and the inlet gas stream are resistively heated to reaction temperature. The reactor temperature is controlled by a thermocouple located in the catalyst bed ∼1 cm from wall and ∼1 cm above distributor plate. The inlet gas temperature is controlled by a thermocouple located in the center of the plenum. During reaction product gases exit the top of the reactor and pass through a cyclone where entrained solids are removed and collected. The vapor then passes through a condenser train. The first three condensers are operated at 0 °C in an ice bath and the following three condensers are operated at −55 °C in a dry ice/acetone bath. The non-condensed vapors exiting the condenser train are collected in a tedlar gas sampling bag for GC/MS and GC/FID analysis. After reaction the condensers are removed and washed with ethanol to collect the liquids condensed during the reaction. The total volume of ethanol/product solution collected is recorded. The solution is then analyzed with GC/MS and GC/FID to identify and quantify the amounts of the various products. For a typical run wood is fed to the reactor for 30 min. After the feed auger is stopped the reactor is purged with 1200 sccm of helium for another 30 min to strip any remaining product from the catalyst.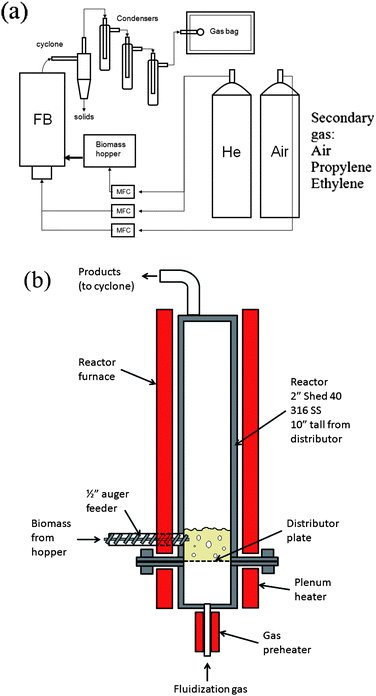 | ||
| Fig. 3 The experimental setup of the fluidized bed reactor system. (a) Schematic of the fluidized bed system and (b) detailed drawing of the reactor. | ||
For the olefin co-feed experiments the secondary gas (T2 in Fig. 1) was switched to either ethylene or propylene and controlled at the desired flow rate. The helium fluidization gas flow rate was adjusted to hold the total inlet gas flow rate constant at 1200 sccm.
After reaction and purge the secondary gas is switched to air to regenerate the catalyst. For a typical run the catalyst was regenerated for approximately three hours to ensure no organic species remained on the catalyst. The combustion effluent during regeneration is passed over a copper catalyst (Sigma Aldrich) held at 250 °C to convert carbon monoxide to carbon dioxide. The carbon dioxide stream then passes over a dryrite trap to remove water vapor. The dry carbon dioxide is collected a preweighted ascarite trap. The total moles of carbon dioxide collected in the trap are equal to the moles of carbon in coke on the catalyst bed.
The gas residence time distribution of the reactor was measured at room temperature by switching the fluidization gas from pure helium to a 2 mol% CO in helium mixture in a step change fashion. After the gas was switched the gas samples were collected every 30 s at the reactor. The concentration of the outlet gas was measured by GC-TCD. Three separate runs were conducted starting at different times (5, 10 and 15 s after the gas switch) to obtain a measurement every 5 seconds.
3.0 Results
3.1 Fluidized bed
 | ||
| Fig. 4 Gas phase aromatic concentrations as a function of time on stream for catalytic fast pyrolysis of pine sawdust. Reaction conditions: pine wood feed at 0.1 WHSV, 1200 sccm He fluidization flow rate, and 600 °C reactor temperature. Key: ◆: benzene, ▲: toluene, ●: xylenes, and ■: total aromatics. | ||
 | ||
| Fig. 5 Normalized gas concentration in the fluidized bed reactor after a step change in inlet concentration. The lines are the calculated concentrations for an ideal PFR (dashed) and an ideal CSTR (solid). | ||
 | ||
| Fig. 6 Carbon yields as a function of biomass WHSV for CFP of pine sawdust. Reaction conditions: ZSM-5 catalyst, 600 °C, 1200 sccm helium fluidization flow rate, and 30 min total reaction time. Key: (a) ◆: aromatics, ○: aromatics + olefins, ■: coke, and □: unidentified, and (b) ▲: CO, △: CO2, ○: methane, and ◊: olefins. | ||
Table 2 gives a detailed carbon yield and selectivity as a function of WHSV. Methane and ethylene are the primary light hydrocarbon species. At low WHSV ethylene is the most abundant light hydrocarbon with a carbon selectivity of 59% followed by methane with a carbon selectivity of 34.2%. At high biomass WHSV methane becomes the dominant light hydrocarbon product (57% carbon selectivity) while ethylene selectivity decreases to 37.0%. The selectivities for toluene and xylene are both strong functions of biomass WHSV. Toluene and xylenes (total of meta, ortho and para) carbon selectivities both decrease with increasing WHSV appreciably from 34.1% to 17.2% and 15.4% to 2.9%, respectively. Benzene and naphthalene show the opposite trend. Benzene increases from 24.8% to 33.4% carbon selectivity while naphthalene increases from 14.9% to 26.1% carbon selectivity as WHSV increases.
| WHSV/h−1 | ||||
|---|---|---|---|---|
| 0.1 | 0.2 | 0.8 | 1.7 | |
| Overall yields | ||||
| Aromatics | 14.0 | 11.0 | 12.1 | 9.5 |
| Olefins | 5.4 | 8.2 | 8.6 | 6.1 |
| Methane | 2.8 | 4.5 | 8.8 | 8.0 |
| Carbon monoxide | 26.2 | 26.3 | 23.1 | 29.9 |
| Carbon dioxide | 9.4 | 8.1 | 4.9 | 5.6 |
| Coke | 36.8 | 30.2 | 26.1 | 19.9 |
| Aromatic selectivity | ||||
| Benzene | 24.8 | 23.1 | 29.1 | 33.4 |
| Toluene | 34.1 | 30 | 21.9 | 17.2 |
| Ethylbenzene | 0.6 | 1.2 | 0.5 | 0.2 |
| m-Xylene and p-xylene | 12.9 | 12 | 5 | 2.6 |
| o-Xylene | 2.5 | 1.9 | 0.8 | 0.3 |
| Styrene | 3.3 | 4.4 | 5.9 | 5.2 |
| Phenol | 1.1 | 4 | 8.1 | 5.1 |
| Indene | 1.4 | 7.1 | 8.9 | 8.4 |
| Benzofuran | 4.3 | 1.6 | 2.1 | 1.4 |
| Naphthalene | 14.9 | 14.7 | 17.7 | 26.1 |
| Light hydrocarbon selectivity | ||||
| Methane | 34.2 | 35.4 | 50.6 | 57 |
| Ethylene | 59.8 | 41 | 41.3 | 37 |
| Propylene | 5.4 | 16.6 | 6.1 | 4.3 |
| Butene | 0.2 | 1.9 | 0.4 | 0.3 |
| Butadiene | 0.4 | 5 | 1.7 | 1.5 |
 | ||
| Fig. 7 Effect of temperature on the carbon yield for CFP of pine sawdust. Reaction conditions: ZSM-5 catalyst, 0.2 wood WHSV, 1200 sccm helium fluidization flow rate, and 30 min total reaction time. Key: (a) ◆: aromatics, ○: aromatics + olefins, ■: coke, and □: unidentified, and (b) ▲: CO, △: CO2, ○: methane, and ◊: olefins. | ||
| Temperature/°C | |||
|---|---|---|---|
| 500 | 600 | 670 | |
| Overall yields | |||
| Aromatics | 7.4 | 11.0 | 9.3 |
| Olefins | 8.8 | 8.2 | 9.2 |
| Methane | 3.1 | 4.5 | 10.9 |
| Carbon monoxide | 14.1 | 26.3 | 30.1 |
| Carbon dioxide | 5.9 | 8.1 | 9.1 |
| Coke | 38.4 | 30.2 | 23.8 |
| Aromatic selectivity | |||
| Benzene | 26.1 | 23.1 | 45.7 |
| Toluene | 41.5 | 30 | 14.6 |
| Ethylbenzene | 3.1 | 1.2 | 0.1 |
| m-Xylene and p-xylene | 8.8 | 12 | 1.1 |
| o-Xylene | 3.8 | 1.9 | 0.1 |
| Styrene | 2.9 | 4.4 | 3.3 |
| Phenol | 4.8 | 4 | 0.5 |
| Indene | 3.2 | 7.1 | 2.5 |
| Benzofuran | 1.2 | 1.6 | 0.4 |
| Naphthalene | 4.5 | 14.7 | 31.7 |
| Light hydrocarbon selectivity | |||
| Methane | 25.7 | 35.4 | 54.1 |
| Ethylene | 45.7 | 41 | 44.8 |
| Propylene | 22.1 | 16.6 | 0.8 |
| Butene | 4 | 1.9 | 0.1 |
| Butadiene | 2.4 | 5 | 0.2 |
Fig. 8 shows the overall yields for the different products from the CFP of wood with propylene co-feed. In Fig. 8 the carbon yield is the single pass yield e.g. it is calculated as the amount of carbon in the given product divided by the total amount of carbon fed to the reactor (wood and olefin). Increasing the amount of propylene co-feed slightly increases the aromatic yield from 11% to 12.4% carbon while the coke yield decreases from 30 to 25%. The yield of carbon dioxide and carbon monoxide also decreases at higher propylene feed concentration. The carbon monoxide and carbon dioxide yields increase from 26.3 to 35.1% carbon and 8.1 to 9.4% carbon, respectively.
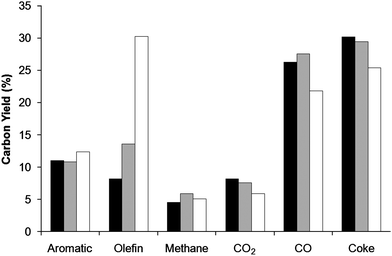 | ||
| Fig. 8 Single pass yields for catalytic fast pyrolysis of pine wood with propylene as a co-feed. The yield based on total carbon fed to the reactor. Reaction conditions: ZSM-5 catalyst, 0.2 wood WHSV, 1200 sccm helium fluidization flow rate, and 30 min total reaction time. Key: black: no co-feed, grey: 0.09 propylene/wood carbon ratio, and white: 0.3 propylene/wood carbon ratio. The aromatics quantified include: benzene, toluene, xylene (all three isomers), ethylbenzene, styrene, indene, phenol and naphthalene and 1-methylnaphthalene. The olefins quantified include: ethylene, propylene, butene and butadiene. | ||
Table 5 shows how the selectivity for the aromatic species changes with the addition of propylene co-feed. In general the aromatic selectivity does not change significantly with the addition of propylene. The selectivity for benzene is the highest (30.4% carbon) at intermediate propylene concentration. The toluene selectivity is highest (33.3%) at the high propylene concentration. At the intermediate propylene concentration toluene exhibits a minimum selectivity of 28% carbon. The total selectivity for xylenes remains relatively constant at ∼12–14% carbon selectivity for low and high propylene concentrations, however, at intermediate concentration far more o-xylene is produced. The benzofuran selectivity is a strong function of propylene concentration and increases from 1.6% to 11.0% carbon as the propylene concentration increases. The selectivity for naphthalene decreases at the higher concentration of propylene.
| Propylene/wood ratio (mol/mol carbon) | |||
|---|---|---|---|
| 0.00 | 0.09 | 0.3 | |
| Overall yields | |||
| Aromatics | 11.0 | 10.8 | 12.4 |
| Olefins | 8.2 | 13.6 | 30.2 |
| Methane | 4.5 | 5.9 | 5.0 |
| Carbon monoxide | 26.3 | 27.6 | 21.8 |
| Carbon dioxide | 8.1 | 7.5 | 5.9 |
| Coke | 30.2 | 29.4 | 25.4 |
| Aromatic selectivity | |||
| Benzene | 23.1 | 30.4 | 24.8 |
| Toluene | 30.0 | 28.0 | 33.3 |
| Ethylbenzene | 1.2 | 0.7 | 1.3 |
| m-Xylene and p-xylene | 12.0 | 6.9 | 11.2 |
| o-Xylene | 1.9 | 4.7 | 2.1 |
| Styrene | 4.4 | 1.2 | 3.6 |
| Phenol | 4.0 | 1.6 | 1.3 |
| Indene | 7.1 | 2.4 | 0.3 |
| Benzofuran | 1.6 | 9.0 | 11.0 |
| Naphthalene | 14.7 | 15.1 | 11.0 |
| Light hydrocarbon selectivity | |||
| Methane | 42.5 | 46.0 | 31.0 |
| Ethylene | 49.2 | 48.7 | 58.5 |
| Propylene | na | na | na |
| Butene | 2.3 | 1.6 | 6.3 |
| Butadiene | 6.0 | 3.7 | 4.2 |
Fig. 9 shows the single pass yield for the various products with ethylene co-fed with wood to the reactor. As shown in Fig. 9 increasing the ethylene in the feed slightly decreases the aromatic and coke yields. The decrease in aromatic yield indicates the ethylene is not reacting to form additional aromatics, which is consistent with the conversion of ethylene in Table 4. The yield of carbon dioxide slightly decreases with increasing ethylene concentration. The carbon monoxide yield goes through a maximum at intermediate ethylene concentration.
 | ||
| Fig. 9 Catalytic fast pyrolysis of pine wood with ethylene as a co-feed. The yield is calculated from the total carbon fed to the reactor. Reaction conditions: ZSM-5 catalyst, 0.2 wood WHSV, 1200 sccm helium fluidization flow rate, and 30 min total reaction time. Key: black: no co-feed, grey: 0.05 ethylene/wood carbon ratio, and white: 0.15 ethylene/wood carbon ratio. The aromatics quantified include: benzene, toluene, xylene (all three isomers), ethylbenzene, styrene, indene, phenol and naphthalene and 1-methylnaphthalene. The olefins quantified include: ethylene, propylene, butene and butadiene. | ||
Table 6 shows how the selectivity for the aromatic species changes with the addition of ethylene co-feed. The aromatic selectivities do not change significantly with ethylene concentration. The benzene selectivity increases from 23.1 to 27.4% as the ethylene concentration increases. The total selectivity for xylenes decreases with increasing ethylene concentration from 13.9 to 9.3% carbon. Similar to the propylene co-feed at intermediate concentrations of olefin the ratio of o-xylene to m-xylene and p-xylene is higher. Benzofuran selectivity is also strong function of ethylene concentration. It increases from 1.6% to 12.0% carbon while going through a maximum of 13.8% at the intermediate concentration. The selectivity for naphthalene (14.7%) is not a strong function of ethylene concentration. The propylene selectivity does not change significantly with ethylene concentration. The selectivity for larger olefins such as butane and butadiene decreases with increasing ethylene concentration.
| Ethylene/wood ratio (mol/mol carbon) | |||
|---|---|---|---|
| 0.00 | 0.05 | 0.15 | |
| Overall yields | |||
| Aromatics | 11.0 | 11.8 | 10.0 |
| Olefins | 8.2 | 15.5 | 26.4 |
| Methane | 4.5 | 5.2 | 4.6 |
| Carbon monoxide | 26.3 | 29.4 | 26.4 |
| Carbon dioxide | 8.1 | 7.2 | 7.2 |
| Coke | 30.2 | 29.4 | 28.7 |
| Aromatics selectivity | |||
| Benzene | 23.1 | 23.6 | 27.4 |
| Toluene | 30 | 24.1 | 26.6 |
| Ethylbenzene | 1.2 | 0.9 | 0.8 |
| m-Xylene and p-xylene | 12 | 7.2 | 7.4 |
| o-Xylene | 1.9 | 4.7 | 1.9 |
| Styrene | 4.4 | 8.7 | 4.5 |
| Phenol | 4 | 1.5 | 1.7 |
| Indene | 7.1 | 2.6 | 3.1 |
| Benzofuran | 1.6 | 13.8 | 12 |
| Naphthalene | 14.7 | 12.8 | 14.5 |
| Light hydrocarbon selectivity | |||
| Methane | 60 | 64.2 | 61.8 |
| Ethylene | na | na | na |
| Propylene | 28.2 | 26.7 | 29.6 |
| Butene | 3.2 | 2.1 | 1.8 |
| Butadiene | 8.5 | 7 | 6.7 |
3.2 Catalyst characterization
To study the stability of the catalyst during CFP the catalyst was subjected to ten reaction/regeneration cycles. For each cycle the reaction was performed with a WHSV of 0.2 h−1 at 600 °C for 30 min followed by a 30 min purge. After reaction and purge the catalyst was regenerated in air for approximately 3 hours. Fig. 10 shows the powder diffraction patterns of the Grace ZSM-5 catalyst before and after 10 reaction/regeneration cycles. The ZSM-5 crystal structure appears to slightly change after 10 reaction–regeneration cycles as the intensities of some of the peaks change. An increase in the intensity of the peak at ∼8 2Θ after reaction indicates an increase in ZSM-5 crystallinity or an increase in the framework Si/Al ratio.33,34 However, it has been previously shown that a decrease in intensity for the peak at 24 2Θ indicates if aluminium is removed from the framework.34,35 As seen in the inset of Fig. 10 the peak at 24 2Θ does not change in intensity, therefore, the increase in Si/Al ratio of the zeolite is small. | ||
| Fig. 10 X-Ray diffraction patterns before reaction (dotted line) and after 10 reaction–regeneration cycles (grey line). Cu anode material, K-α1 wavelength = 1.540598 Å, K-α2 wavelength = 1.544426 Å, ratio K-α2/K-α1 = 0.5, and fixed divergence slit at 0.10 mm. | ||
To qualitatively determine the loss of catalyst fines and attrition SEM imaging was used before and after reaction. The SEM images of the fresh catalyst (a) and the catalyst after ten reaction/regeneration cycles (b) are shown in Fig. 11. The average particle size was measured using ImageJ image processing and analysis software. The average particle size was found to increase from 45 to 63 µm after 10 reaction–regeneration cycles. The increase in particle size suggests there is a loss of catalyst fines. This is probably because of loss of catalyst from the fluidized bed reactor due to entrainment. The entrained catalyst was not recycled from the cyclone back to the reactor. The image of the used catalyst also shows some signs of physical damage. More broken pieces of catalyst can be seen in Fig. 11b compared to Fig. 11a.
 | ||
| Fig. 11 SEM images of the fluidized bed catalyst before (a) and after 10 reaction–regeneration cycles (b). | ||
The DRIFT spectra of adsorbed ammonia at 100 °C are shown in Fig. 12. The assignments of the bands and relative areas are reported in Table 7. It can be seen that the area of the bands at 3675 and 3610 cm−1 which correspond to Lewis and Brønsted acids do not change much after 10 reaction–regeneration cycles. The ratio of Brønsted to Lewis acid sites increases slightly from 1.4 to 1.6 after the repeated reaction–regeneration. The disappearance of the band at 3745 cm−1 indicates that the number of surface hydroxyl groups decreases after the reaction–regeneration cycles.
 | ||
| Fig. 12 DRIFT spectra of adsorbed ammonia on the fluidized bed catalyst at 500 °C. Fresh is the catalyst as-received after calcining and spent is the catalyst after 10 reaction–regeneration cycles. | ||
In addition to the distribution of acid sites the total number of acid sites was measured using ammonia temperature programmed desorption (TPD). The TPD curves for the fresh and spent catalysts are shown in Fig. 13. It can be seen that there are two peaks with centers at ∼275 °C and ∼475 °C. The low temperature peak corresponds to the weakly bound ammonia on non-framework Lewis acid sites whereas the high temperature peak corresponds to the more strongly bound ammonia on Brønsted acid sites.36 As reported in Table 8 the total acidity of the catalyst decreases after the 10 reaction–regeneration cycles. From the TPD curve it appears that the loss in acidity is due to a decrease of the low temperature peak intensity as the high temperature peak does not change much.
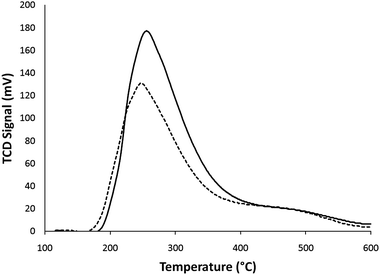 | ||
| Fig. 13 Temperature programmed desorption of ammonia for the fresh (solid line) and spent (dotted line) catalysts. | ||
To determine whether metals found in the biomass are deposited on the catalyst during reaction the spent catalyst was subjected to ICP-EOS to measure the elements present. It can be seen that ppm levels of four common biomass metals are found in the spent catalyst. It can also be seen that the bulk weight percentage of silicon and aluminium stays relatively the same after the 10 reaction–regeneration cycles. The ratio of silicon to aluminium is also constant at a value of 2. The other primary element in the catalyst is phosphorous. The weight percentage of phosphorous in the catalyst also only changes slightly from 4.75 to 4.5 wt% after ten reaction–regeneration cycles. Typically spray dried catalysts contain above 40 wt% zeolite, 3–15 wt% phosphorus (P2O5), 15–45% kaolin (Al2Si2O5(OH)4) and above 10 wt% alumina.37 The low Si/Al ratio is probably due to other additives such as kaolin and alumina as ZSM-5 catalysts typically have a Si/Al ratio above 10 (Table 9).
| Fresh | Spent | |
|---|---|---|
| Element present | ||
| Aluminium/wt% | 13.4 | 13.2 |
| Silicon/wt% | 26.2 | 27.5 |
| Phosphorous/wt% | 4.75 | 4.5 |
| Calcium (ppm) | 0 | 642 |
| Potassium (ppm) | 0 | 812 |
| Magnesium (ppm) | 0 | 308 |
| Manganese (ppm) | 0 | 88 |
3.3 Pyroprobe
Since the powdered Zeolyst catalyst sample is unsuitable for use in the fluidized bed reactor the pyroprobe was used to compare the zeolyst catalyst with the spray dried catalyst. The low density and small particle size of the Zeolyst catalyst would require far lower fluidization gas flows and a comparison to the spray dried catalyst using the same reaction conditions would be impossible. In the pyroprobe reactor the same reaction conditions can be used for both catalyst samples. The pyroprobe reactor is a semi-batch reactor where small samples of biomass and catalyst are mixed together. The pyroprobe reactor is then rapidly heated and the products are analyzed by GCMS. The pyroprobe reactor can screen large numbers of catalysts in a short time with each reaction taking less than 30 minutes to complete. Fig. 14 shows the performance of the two catalysts for the conversion of wood and furan.13 It can be seen that the aromatic yield of 18% carbon from wood with the Grace catalyst is higher than the best yield of 14% aromatic yield obtained in the fluidized bed reactor. Using the pure ZSM-5 Zeolyst catalyst the aromatic yield from wood increases to 24% carbon. This demonstrates that the pure ZSM-5 catalyst is more selective than the spray dried catalyst. The coke yield was higher on the Grace catalyst (42%) compared to the pure ZSM-5 (28%) catalyst. The carbon dioxide yields from the two catalysts are quite similar while the carbon monoxide yield was higher for the Zeolyst catalyst. For the reaction of furan with the Zeolyst ZSM-5 the aromatic yield was 33% carbon. The coke yield for the furan feed was 37% carbon. These results illustrate that furan has similar yields for CFP as wood, and therefore is a good probe molecule. Furthermore, furan has been shown to be an important reaction intermediate in CFP.13 | ||
| Fig. 14 Product yields for the pyrolysis of wood and furan with the two different catalysts. Reaction conditions: catalyst to feed weight ratio 19, nominal heating rate 1000 °C s−1, reaction temperature 600 °C, and reaction time 240 s. Key: aromatics: black, carbon monoxide: white, carbon dioxide: dark grey, and coke (grey). | ||
As shown in Table 10 the aromatic selectivity for the pyrolysis of wood is different for the two catalyst tested. The Grace catalyst produced more naphthalene, indene and phenol. The Zeolyst catalyst produced more of the monocyclic aromatics. The aromatic selectivity for the conversion of furan with the Zeolyst catalyst is quite different from wood. The selectivity for benzene and naphthalene is higher for furan than wood. The toluene and xylene selectivities were lower for furan than for wood. These results demonstrate that the catalyst tested in the fluidized bed reactor is far from the optimal catalyst for CFP.
| Feed/catalyst combination | |||
|---|---|---|---|
| Wood/Zeolyst ZSM-5 | Wood/Grace ZSM-5 | Furan/Zeolyst ZSM-5 | |
| Overall yields | |||
| Aromatics | 24.1 | 17.7 | 33.0 |
| Carbon monoxide | 21.3 | 13.2 | 17.6 |
| Carbon dioxide | 6.8 | 6.0 | 6.3 |
| Coke | 28.3 | 42.2 | 33.6 |
| Aromatic selectivity | |||
| Benzene | 9.7 | 5.2 | 14.0 |
| Toluene | 19.5 | 14.5 | 15.8 |
| Xylene + ethylbenzene | 20.8 | 19.0 | 8.0 |
| Trimethylbenzene + ethyl-methyl-benzene | 6.2 | 4.0 | 0.4 |
| Phenol | 0.4 | 5.3 | 0.0 |
| Benzofuran | 0.0 | 0.0 | 1.6 |
| Indene | 4.2 | 6.2 | 3.3 |
| Naphthalene | 39.3 | 45.8 | 57.0 |
3.4 Fixed bed
 | ||
| Fig. 15 Carbon yield for furan conversion over ZSM-5. Reaction conditions: Zeolyst ZSM-5 catalyst, 204 sccm helium flow rate, 600 °C reactor temperature, and 4.5 min total reaction time. Yield is defined as moles of carbon in the product divided by moles of furan carbon converted. Key: ◆: aromatics, ■: coke, □: unidentified, ▲: CO, △: CO2, ○: methane, and ◊: olefins. | ||
In the fixed bed the aromatic selectivity is also a function of WHSV. The selectivity for toluene and xylene decreases from 37.0% to 25.7% carbon and 10.3 to 0.0% carbon, respectively. Unlike the fluidized bed the selectivity for benzene decreases over the range tested from 37.0% to 27.2% carbon. The selectivity for the intermediate size aromatics indene and benzofuran changes the most over the range tested. Indene increases from 7.5% to 24.2% while benzofuran increases from 0.6% to 12.2%. The selectivity for naphthalene is relatively constant over the range (Table 11).
| WHSV/h−1 | |||
|---|---|---|---|
| 1.9 | 10.4 | 98.4 | |
| Furan conversion | 96.9 | 43.3 | 17.2 |
| Overall yields | |||
| Aromatics | 26.7 | 24.3 | 7.2 |
| Olefins | 17.5 | 16.4 | 4.3 |
| Carbon monoxide | 17.0 | 17.1 | 3.3 |
| Carbon dioxide | 3.5 | 1.8 | 0.0 |
| Coke | 34.5 | 32.5 | 10.3 |
| Aromatic selectivity | |||
| Benzene | 37.0 | 33.5 | 27.2 |
| Toluene | 37.0 | 30.0 | 25.7 |
| Ethylbenzene | 0.0 | 0.0 | 0.0 |
| Xylenes | 10.3 | 5.4 | 0.0 |
| Styrene | 5.5 | 9.2 | 7.7 |
| Indene | 7.5 | 13.7 | 24.2 |
| Benzofuran | 0.6 | 3.6 | 12.2 |
| Naphthalene | 2.1 | 4.6 | 2.9 |
| Light hydrocarbon selectivity | |||
| Ethylene | 54.0 | 50.2 | 61.8 |
| Propylene | 41.3 | 45.4 | 38.2 |
| Butene | 4.7 | 4.4 | 0.0 |
 | ||
| Fig. 16 Conversion of furan as a function of temperature over a zeolite catalyst. Reaction conditions: Zeolyst ZSM-5 catalyst, 204 sccm helium flow rate, 10.4 WHSV, and 4.5 min total reaction time. Carbon yield defined as moles of carbon in the product divided by moles of furan converted. Key: ◆: aromatics, ■: coke, □: unidentified, ▲: CO, △: CO2, ○: methane, and ◊: olefins. | ||
The aromatic selectivity is also a strong function of temperature. The selectivity for benzene increases from 14.9% to 39.7% carbon as the temperature increases. The selectivity for toluene increases with temperature from 17.4% to 29.0% carbon. Xylene and naphthalene go through a maximum selectivity at 500 °C and 550 °C, respectively. At low temperature benzofuran is the most abundant product with a selectivity of 51.5 carbon percent, however, the selectivity decreases to 11.3% when the temperature is increased to 650 °C (Table 12).
| Temperature/°C | |||||
|---|---|---|---|---|---|
| 450 | 500 | 550 | 600 | 650 | |
| Overall yields | |||||
| Aromatics | 18.8 | 20.5 | 21.3 | 24.3 | 20.9 |
| Olefins | 5.0 | 7.5 | 10.8 | 16.4 | 19.4 |
| Carbon monoxide | 8.8 | 12.7 | 14.2 | 17.1 | 18.0 |
| Carbon dioxide | 0.5 | 1.1 | 1.5 | 1.8 | 1.2 |
| Coke | 22.1 | 26.6 | 18.5 | 32.5 | 17.5 |
| Aromatic selectivity | |||||
| Benzene | 14.9 | 19.1 | 26.0 | 33.5 | 39.7 |
| Toluene | 17.4 | 24.2 | 28.5 | 30.0 | 29.0 |
| Ethylbenzene | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Xylenes | 5.0 | 6.6 | 5.9 | 5.4 | 4.9 |
| Styrene | 1.8 | 5.2 | 7.7 | 9.2 | 7.4 |
| Indene | 9.5 | 13.3 | 14.0 | 13.7 | 4.1 |
| Benzofuran | 51.5 | 29.6 | 13.1 | 3.6 | 11.3 |
| Naphthalene | 0.0 | 2.1 | 4.7 | 4.6 | 3.7 |
| Light hydrocarbon selectivity | |||||
| Ethylene | 61.6 | 56.8 | 51.5 | 50.2 | 55.5 |
| Propylene | 38.4 | 43.2 | 47.6 | 45.4 | 38.9 |
| Butene | 0.0 | 0.0 | 0.9 | 4.4 | 5.6 |
Fig. 17 shows the single pass yields of the various products as a function of ethylene co-feed amount. The single pass yield for 2 mol% ethylene without furan feed is also shown. The yield of aromatics increases slightly at the intermediate ethylene amount then decreases at higher ethylene concentration. This is from the low reactivity of ethylene as more carbon is being fed to the reactor without greatly increasing the total amount of aromatics produced.
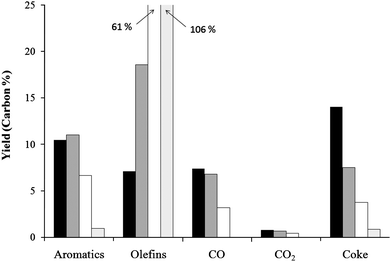 | ||
| Fig. 17 Yields for the reaction of furan with ethylene co-feed over ZSM-5. The yield is calculated from the total carbon fed to the reactor. Key: black: no co-feed, grey: 0.13 ethylene/furan carbon ratio, white: 1.24 ethylene/furan carbon ratio, and light gray: 2% ethylene only. | ||
The selectivity for the various aromatic products is also a function of the amount of ethylene in the co-feed as shown in Table 14. The selectivity for both toluene and xylenes increases at the higher ethylene feed amounts. Benzene, benzofuran and naphthalene all decrease slightly from no co-feed to high ethylene co-feed. However, benzene goes through a minimum at intermediate ethylene feed while benzofuran and naphthalene go through a maximum. The primary olefin produced other than ethylene is propylene with a selectivity of 91.1% at no ethylene co-feed. At the highest ethylene feed amount the propylene selectivity decreases to 87.1% carbon while butene increases from 8.9% to 12.9% carbon.
| Ethylene/furan ratio (mol/mol carbon) | ||||
|---|---|---|---|---|
| 0.00 | 0.13 | 1.24 | 2% Ethylene only | |
| Overall yields | ||||
| Aromatics | 10.5 | 11.0 | 6.6 | 1.0 |
| Olefins | 7.1 | 18.6 | 61.4 | 106.1 |
| Carbon monoxide | 7.4 | 6.8 | 3.2 | 0.0 |
| Carbon dioxide | 0.8 | 0.7 | 0.4 | 0.0 |
| Coke | 14.1 | 7.5 | 3.7 | 0.8 |
| Aromatic selectivity | ||||
| Benzene | 33.5 | 30.2 | 31.3 | 46.6 |
| Toluene | 30.0 | 29.4 | 33.4 | 25.1 |
| Xylenes | 5.4 | 6.6 | 9.1 | 14.7 |
| Styrene | 9.2 | 9.4 | 8.1 | 0.0 |
| Indene | 13.7 | 15.1 | 11.5 | 13.6 |
| Benzofuran | 3.6 | 4.4 | 2.8 | 0.0 |
| Naphthalene | 4.6 | 4.9 | 3.7 | 0.0 |
| Light hydrocarbon selectivity | ||||
| Ethylene | na | na | na | na |
| Propylene | 91.1 | 91.5 | 87.1 | 56.1 |
| Butene | 8.9 | 8.5 | 12.9 | 43.9 |
Fig. 18 shows the single pass yields of the various products for different propylene co-feed amount. The single pass yield for 2 mol% propylene without furan feed is also shown. The yield of aromatics increases from 10.5% to 16.3% carbon. A synergistic effect can clearly be seen in Fig. 17 as this aromatic increase is higher than the sum of the aromatic yields from the furan and 2 mol% propylene. Like ethylene the propylene co-feed decreases the coke yield. The coke yield is decreased from 14.0% to 3.7% carbon.
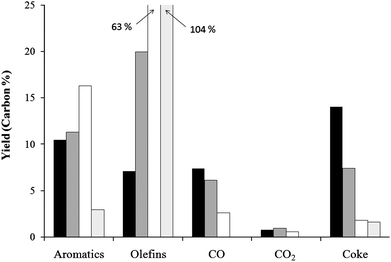 | ||
| Fig. 18 Yields for the reaction of furan with propylene co-feed over ZSM-5. The yield is calculated from the total carbon fed to the reactor. Key: black: no co-feed, dark grey: 0.18 propylene/furan carbon ratio, white: 1.83 propylene/furan carbon ratio, and light gray: 2% propylene only. | ||
As shown in Table 15propylene co-feed affects the selectivity of aromatics more than the ethylene co-feed. The selectivity for benzene decreases from 33.5% to 16.2% carbon with increasing olefin amount. Interestingly for the propylene run without furan feed benzene is the most selective product at 39.1% carbon selectivity. Toluene selectivity doubles from 30.0% to 60.4% carbon at the highest propylene/furan ratio. Xylenes also increase with propylene feed from 5.4% to 15.2%. Styrene, indene, benzofuran and naphthalene selectivity all decrease with propylene feed. The propylene co-feed also has an effect on the other olefins. The ethylene selectivity decreases from 91.9% to 64.8% carbon while butene increases from 8.1% to 35.2% carbon.
| Propylene/furan ratio (mol/mol carbon) | ||||
|---|---|---|---|---|
| 0.00 | 0.18 | 1.83 | 2% Propylene only | |
| Overall yields | ||||
| Aromatics | 10.5 | 11.3 | 16.3 | 3.0 |
| Olefins | 7.1 | 19.9 | 62.9 | 104.2 |
| Carbon monoxide | 7.4 | 6.1 | 2.6 | 0.0 |
| Carbon dioxide | 0.8 | 1.0 | 0.6 | 0.0 |
| Coke | 14.1 | 7.4 | 1.8 | 1.6 |
| Aromatic selectivity | ||||
| Benzene | 33.5 | 28.7 | 16.2 | 39.1 |
| Toluene | 30.0 | 39.9 | 60.4 | 35.4 |
| Xylenes | 5.4 | 8.1 | 15.2 | 14.7 |
| Styrene | 9.2 | 7.7 | 4.5 | 1.4 |
| Indene | 13.7 | 9.7 | 2.3 | 6.0 |
| Benzofuran | 3.6 | 2.1 | 0.6 | 0.0 |
| Naphthalene | 4.6 | 3.8 | 0.9 | 3.4 |
| Light hydrocarbon selectivity | ||||
| Ethylene | 91.9 | 84.2 | 64.8 | 51.4 |
| Propylene | na | na | na | Na |
| Butene | 8.1 | 15.8 | 35.2 | 48.6 |
4.0 Discussion
4.1 Reactor design
In this paper three different reactor configurations and two different feedstocks were tested. Fig. 19 shows the optimized yields for the three reactors. For the continuous reactors the lowest WHSV run is shown. Both feeds showed a higher yield of aromatics in the pyroprobe reactor than the continuous reactors with the same catalyst. The coke yield is also higher in the pyroprobe reactor. No olefins were detected in the pyroprobe reactor. Furthermore, larger amounts of naphthalenes are observed in the pyroprobe reactor. The olefins plus aromatic yields are higher in the continuous reactors than the pyroprobe reactor. The differences in the three reactors may arise from differences in mass transfer. In the continuous reactors there is a high gas flux through the catalyst bed while the pyroprobe reactor has no gas flow though the bed. In the pyroprobe reactor the gas residence time and concentration are likely much higher. With a long gas residence time the rate of naphthalene formation could be higher as it is probably formed by secondary reactions.18 The absence of olefins in the product from the pyroprobe could also be linked to low rates of mass transfer in the pyroprobe as olefins can oligomerize to form aromatics.19,22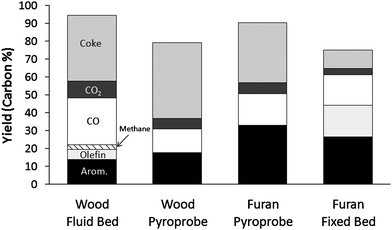 | ||
| Fig. 19 Comparison of all three reactors with optimized reaction conditions. Key: aromatics: black, carbon monoxide: white, carbon dioxide: dark grey, coke: medium grey, olefins: dark grey, and methane: hatched lines. | ||
Aside from reactor configuration, temperature and weight hourly space velocity have the largest effect on aromatic yield and selectivity. It may be advantageous to operate at temperatures below 600 °C as this temperature maximizes the monocyclic aromatics. Higher temperatures shift the selectivity toward naphthalenes. The olefin selectivity exiting the reactor is a strong function of temperature. In the fluidized bed at temperatures below 600 °C propylene is selectively produced and could be recycled to form more aromatics. In the fixed bed with furan the maximum selectivity for propylene is at 550 °C. Operation at low temperature also decreases the amount of methane generated during wood pyrolysis. The methane is likely from the lignin portion of the pinewood as only trace amounts of methane were measured during the conversion of furan (Table 16).
| Feed/reactor combination | ||||
|---|---|---|---|---|
| Wood/fluidized bed | Wood/pyroprobe | Furan/pyroprobe | Furan/fixed bed | |
| Overall yields | ||||
| Aromatics | 14.0 | 17.7 | 33.0 | 26.7 |
| Olefins | 5.4 | 0.0 | 0.0 | 17.6 |
| Methane | 2.8 | 0.0 | 0.0 | 0.0 |
| Carbon monoxide | 26.2 | 13.2 | 17.6 | 17.1 |
| Carbon dioxide | 9.4 | 6.0 | 6.3 | 3.5 |
| Coke | 36.8 | 42.2 | 33.6 | 10.3 |
| Aromatic selectivity | ||||
| Benzene | 24.8 | 5.2 | 14.0 | 37.0 |
| Toluene | 34.1 | 14.5 | 15.8 | 37.0 |
| Xylene + ethylbenzene | 19.4 | 19.0 | 8.0 | 15.8 |
| Trimethylbenzene + ethyl-methyl-benzene | 0.0 | 4.0 | 0.4 | 0.0 |
| Phenol | 1.1 | 5.3 | 0.0 | 0.0 |
| Benzofuran | 4.2 | 0.0 | 1.6 | 0.6 |
| Indene | 1.4 | 6.2 | 3.3 | 7.5 |
| Naphthalene | 14.9 | 45.8 | 57.0 | 2.1 |
In addition to temperature, the biomass WHSV can be used to maximize the yield of toluene and xylene. As seen in Table 2 both of these aromatics have high selectivity at low WHSV. Furthermore, when using wood as the feedstock the selectivity for the undesired naphthalene and methane decreases at low WHSV. If the objective is to use the aromatics as a gasoline additive toluene and xylene would be the best aromatics to produce as they are higher octane than benzene and naphthalene.38 Additionally EPA regulations limit benzene to 0.8 vol% in gasoline while the other aromatics can make up to 25% of the total volume.39 However, benzene is more valuable than toluene due to its use in the chemical industry.
The major competing reaction to the formation of aromatics is the formation of coke. The time on stream study shows that catalyst activity goes through a maximum. This initial increase in aromatic concentration is likely due to the formation of the hydrocarbon pool within the zeolite. It is possible that this hydrocarbon pool acts as a catalyst to selectively produce the aromatics. When the maximum rate is obtained the hydrocarbon pool is likely fully formed within the zeolite, however, at the same time the coking of the catalyst begins to deactivate the activity. Therefore the aromatic concentration decreases in the outlet gas. To be industrially feasible fresh catalyst would need to be continuously feed to the reactor while spent catalyst is withdrawn and regenerated in a separate vessel. The process heat from regeneration could be used to provide energy for the pyrolysis reactor.
In addition to reversible catalyst deactivation by coking irreversible deactivation by loss of zeolite crystal structure, active sites and attrition of the catalyst particles could occur. Several researchers have shown that ZSM-5 is susceptible to loss of acid sites by dealumination under steam treatment.40–43 It is likely that the water concentration in the reactor is too low to see this kind of deactivation as the XRD data before and after reaction show that the crystal structure of the zeolite is relatively unchanged. The TPD data show the total acidity of the composite catalyst decreases with repeated reaction/regeneration cycles, however, this loss in acidity appears to be from a loss of the weak non-framework Lewis acid sites in the non-zeolite components of the catalyst (such as alumina).36 The actual zeolite acid sites are not likely lost as the high temperature TPD peak and the ratio of Brønsted to Lewis acid sites in the zeolite measured by DRIFTS remain constant. From the ICP-OES the metals, calcium, potassium, magnesium and manganese, are deposited on the catalyst after the repeated reaction–regeneration cycles. The deactivation of zeolite catalyst from these metals has not been documented in the literature, however, other metals such as nickel and vanadium have been well studied.44–47Vanadium effects the crystallinity of the catalyst while Ni promotes coke deposition on the catalyst. For the time on stream used in this study no change in the catalyst activity was observed. However, ppm levels of metals are present on the catalyst after 10 reaction/regeneration cycles. This accumulation of metals on the catalyst could affect catalyst stability for longer times on stream. The long term catalyst deactivation and the design of more stable catalysts is an important area for future research.
The surface hydroxyl groups appear to be removed after repeated reaction–regeneration cycles. From the SEM images the spent catalyst shows signs of particle attrition and loss of fines. According to Werther and Reppenhagen48 the main source of attrition in low superficial velocity (<0.55 m s−1) fluidized bed systems is from the gas jets near the distributor plate and from bubbling within the bed. At higher gas velocities (>0.55 m s−1) the main source of attrition comes from the cyclone. Future reactor design should focus on optimization for catalyst lifetime as well as aromatic yield.
4.2 Olefin recycle
One of the major products from catalytic fast pyrolysis is olefins. For wood the highest olefin yield is about 8% carbon and for furan the highest olefin yield is about 17% carbon. It would be desirable to recycle the product olefins to increase the overall aromatic yield of the process. Propylene is easier to convert into aromatics than ethylene because the carbocation of propylene is more stable than that of ethylene.49 For the conversion of methanol to aromatics over ZSM-5 it has been proposed that ethylene is an end product formed from the hydrocarbon pool concurrently with aromatics. Propylene is suspected to be an intermediate in a separate cycle which forms higher alkenes.22 These higher alkenes ultimately oligomerize to form aromatics.19,22 The results from Table 2 also suggest that propylene is an intermediate as it exhibits a maximum yield at intermediate biomass WHSV.The feasibly of recycling olefins to the reactor can be assessed by simple mass balance on the model system shown in Fig. 20. The wood (labeled biomass in Stream 1) is mixed with a recycle stream (Stream 3) containing the olefins, CO, and CO2, and fed into the fluidized bed reactor. Inside the reactor the wood (dry basis) can either react to form aromatics and olefins by reactions (1) and (2), respectively. The olefins in the reactor can be converted to additional aromatics by eqn (3).
| C3.8H5.8O2.7 → 0.36C7H8 + 1.45H2O + 1.25CO | (1) |
| C3.8H5.8O2.7 → 0.67C3H6 + 0.9H2O + 1.8CO | (2) |
| C3H6 → 3/7C7H8 + 9/7H2 | (3) |
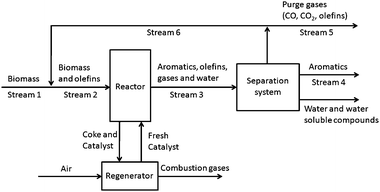 | ||
| Fig. 20 Block flow diagram for aromatic production by catalytic fast pyrolysis. | ||
The balance of biomass not converted into aromatics or olefins is converted into coke and gasses. The spent coked catalyst is then sent to a regenerator and regenerated by burning the coke in a secondary regeneration reactor. Most likely the catalyst recirculation is adjusted to control the temperature of the reactor and regenerator.23 In our system the coke yield is quite high and heat removal from the regenerator may be necessary to avoid high temperatures in the regenerator. The excess heat could be utilized elsewhere in the process. The product stream from the reactor (Stream 3) is separated into the condensable aromatic product (Stream 4), water and water soluble compounds, and non-condensable olefins and gases. The separation system would include a condenser system that removes condensable compounds from the recyclable gases. The liquified product would contain a mixture of water, aromatics and water soluble compounds. The aromatic product would be decanted and further refined. The water and water soluble products would go to waste water treatment. Depending on the reaction conditions, a heavy oxygenated tar product may also be produced. As shown in Fig. 6 and 7, a large amount of unidentified products are produced at low temperature and at high wood WHSV. Under these conditions the liquid product may require a more intensive separation. From the separation system the olefins are then recycled to the reactor with a molar recycle ratio defined as moles of olefin in the recycle (Stream 6) divided by moles of olefin in the purge stream (Stream 5). The purge stream is necessary to remove the CO and CO2 and avoid accumulation of any other non-reactive species in the system.
Fig. 21 shows the effect of adjusting the olefin conversion and recycle ratio for CFP. Each line in Fig. 21 corresponds to a different extent of reaction for eqn (3). The extent of reactions for eqn (1) and (2) was both fixed at 0.17 to match the experimental yield for olefins and aromatics at zero olefin co-feed in the fluidized bed reactor. As shown in Fig. 21 the yield of aromatics increases with increasing the recycle ratio and also increasing the extent of reaction for reaction (3). It can be seen that using recycle ratios in excess of 10 and having high extents of reaction for reaction (3) could lead to a two fold increase in aromatic yield for the system.
 | ||
| Fig. 21 Aromatic yield as a function of recycle ratio for the model process depicted in Fig. 19. Solid lines are drawn for various extents of olefin reaction (eqn (3) above). The extents of reaction plotted are: 0.1, 0.2, 0.3, 0.4 and 0.5. | ||
Recycling the olefins back into the reactor may also allow operation of the fluidized bed reactor at higher space velocities. As shown in the results from Section 3.4.3 there is higher conversion of intermediate furans in the presence of the olefin co-feed. This suggests that co-feeding of olefins at higher biomass WHSV could increase the conversion which otherwise would be low at those conditions.
Shown in Fig. 22 is the volume yield of aromatics per metric ton of feed as a function of the theoretical yield. The volume of aromatics produced at 100% theoretical yield was calculated from eqn (4) which assumes that the molar outlet ratio of CO to CO2 in the product gas is 1.6 to 1. The theoretical yield is a function of the ratio of CO to CO2 produced. Changing the ratio of CO to CO2 changes the stoichiometry of eqn (4). For example, if only CO is produced the theoretical carbon yield of aromatics from wood is 66.3%. If only CO2 is produced, the theoretical carbon yield is 79.2%.
| C3.8H5.8O2.7 → 0.40C7H8 + 1.3H2O + 0.63CO + 0.393CO2 | (4) |
 | ||
| Fig. 22 Volume of aromatics that could be produced from one ton of feed for various yields. | ||
The aromatic yield in our fluidized bed reactor was 17% higher than the aromatic yield obtained by Olazar et al.2 in a spouted bed reactor. If the product olefins are recycled then the yield of aromatics can be increased to 23 gallons per ton. The aromatic yield from wood in the pyroprobe is higher than either of the fluidized bed results. This shows that there is still potential for the optimization of the fluidized bed reactor and the fluidized bed catalyst. The results from Fig. 14 show that the pure zeolite catalyst performs better than the spray dried composite catalyst suggesting further improvements in this process can come by further catalyst improvement. As shown in Fig. 22, the aromatic yield for cellulose CFP is much higher than with wood (∼30 gal ton−1). This suggests that another option for increasing aromatic yield in CFP is to optimize the biomass feedstock by increasing the amount of cellulose and hemicellulose and decreasing the amount of lignin. This result also suggests that the lignin content of the wood decreases the yield of aromatics as we have previously reported for the CFP of maple wood and maple wood with lignin removed.50 In addition, a recent international study involving 14 laboratories concluded that lignin pyrolyses differently than whole biomass and current reactor designs are not sufficient to pyrolyze lignin by itself.51 It has also been shown that for non-catalytic pyrolysis the type of the feedstock can greatly affect the composition of the primary pyrolysis vapors.52 Another way to further increase aromatic yield is to inhibit coke forming reactions as it has been previously shown that coke formation and aromatic production are competing reactions.13 On an energy basis the yield of aromatics from CFP in our current fluidized bed reactor is about half of the projected yield of other biomass conversion technologies such as fermentation and gasification. Ethanol production from wood viahydrolysis and fermentation can yield 85 gal ton−1.53 On an energy basis this volume of fuel yields 7.5 GJ ton−1. It has been projected that up to 56 gal ton−1 liquid alkanes can be produced via Fischer–Tropsch synthesis.53 The energy yield for this volume of fuel is about 8 GJ ton−1. However, it should be noted that large amounts of resources have been devoted to optimize hydrolysis/fermentation technologies and Fischer–Tropsch synthesis whereas few resources have been devoted to the study of CFP.53 Furthermore, CFP is only 25–35% of the theoretical yield today. There is no thermodynamic limitation to the yield that we have obtained as these reactions are thermodynamically favorable. It is likely that advances in catalysis combined with reaction engineering studies to design fluidized bed reactors that are optimized for CFP technology will allow us to obtain energy yields that are comparable to other biomass conversion technologies.
Conclusions
The general conclusion from this study is that aromatics and olefins can be produced directly from wood in a continuous fashion in a fluidized bed reactor that contains zeolite catalysts. The olefins that are produced can be recycled to the reactor to produce additional aromatics. Propylene is more reactive than ethylene leading to a higher conversion of feed and a higher yield of aromatics when it is co-fed to the reactor. Temperature and WHSV can be used to adjust both the yield and selectivity for the aromatic products. When wood is reacted at low space velocities the more valuable monocyclic aromatics are produced and the formation of lower value polycyclic aromatics is inhibited. The more valuable aromatics are also favored at lower temperature. Lowering the temperature also decreases the amount of methane produced. Mineral impurities from the biomass can be found on the zeolite catalyst after the reaction. However, the concentration of acid sites on the zeolite did not change after exposure to the mineral impurities. The spray dried composite catalyst is not as selective to aromatics as the pure ZSM-5 catalyst suggesting that modifying the properties of the spray dried catalyst would increase the aromatic yield. Of the three reactors tested the batch pyroprobe reactor produces the most aromatics. The pyroprobe reactor also produces more naphthalene and does not produce olefins. The combined yield of aromatic plus olefin products is higher in the fixed and fluidized bed reactors with the same feeds and catalysts.Acknowledgements
This work was funded by an NSF CAREER grant, the Catalysis Center for Energy Innovation and by the Defense Advanced Research Project Agency through the Defense Science Office Cooperative Agreement 30 W911NF-09-2-0010 (Surf-Cat: Catalysts for production of JP-8 range molecules from lignocellulosic Biomass. Approved for Public Release, Distribution Unlimited). The views, opinions, and findings contained in this article are those of the author and should not be interpreted as representing the 35 official views or policies, either expressed or implied, of the Defense Advanced Research Projects Agency or the Department of Defense. The Catalysis Center for Energy Innovation is part of an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001004.References
- A. Aho, N. Kumar, K. Eraenen, T. Salmi, M. Hupa and D. Y. Murzin, Fuel, 2008, 87, 2493 CrossRef CAS.
- M. Olazar, R. Aguado, J. Bilbao and A. Barona, AIChE J., 2000, 46, 1025 CrossRef CAS.
- A. A. Lappas, M. C. Samolada, D. K. Iatridis, S. S. Voutetakis and I. A. Vasalos, Fuel, 2002, 81, 2087 CrossRef CAS.
- R. French and S. Czernik, Fuel Process. Technol., 2010, 91, 25 CrossRef CAS.
- T. R. Carlson, G. A. Tompsett, W. C. Conner and G. W. Huber, Top. Catal., 2009, 52, 241 CrossRef CAS.
- T. R. Carlson, T. R. Vispute and G. W. Huber, ChemSusChem, 2008, 1, 397 CrossRef CAS.
- A. Pattiya, J. O. Titiloye and A. V. Bridgwater, J. Anal. Appl. Pyrolysis, 2008, 81, 72 CrossRef CAS.
- R. J. Evans and T. A. Milne, Energy Fuels, 1987, 1, 123 CrossRef CAS.
- J. B. Paine, Y. B. Pithawalla and J. D. Naworal, J. Anal. Appl. Pyrolysis, 2008, 83, 37 CrossRef CAS.
- J. B. Paine, Y. B. Pithawalla and J. D. Naworal, J. Anal. Appl. Pyrolysis, 2008, 82, 10 CrossRef CAS.
- J. B. Paine, Y. B. Pithawalla and J. D. Naworal, J. Anal. Appl. Pyrolysis, 2008, 82, 42 CrossRef CAS.
- Y. C. Lin, J. Cho, G. A. Tompsett, P. R. Westmoreland and G. W. Huber, J. Phys. Chem. C, 2009, 113, 20097 CrossRef CAS.
- T. R. Carlson, J. Jae, Y.-C. Lin, G. A. Tompsett and G. W. Huber, J. Catal., 2010, 270, 110 CrossRef CAS.
- J. Cho, J. M. Davis and G. W. Huber, ChemSusChem, 2010, 3 DOI:10.1002/cssc.201000119.
- P. R. Patwardhan, J. A. Satrio, R. C. Brown and B. H. Shanks, J. Anal. Appl. Pyrolysis, 2009, 86, 323 CrossRef CAS.
- T. Q. Hoang, X. L. Zhu, T. Sooknoi, D. E. Resasco and R. G. Mallinson, J. Catal., 2010, 271, 201 CrossRef CAS.
- A. Corma, G. W. Huber, L. Sauvanaud and P. O'Connor, J. Catal., 2007, 247, 307 CrossRef CAS.
- T. R. Carlson, J. Jae and G. W. Huber, ChemCatChem, 2009, 1, 107 Search PubMed.
- J. F. Haw, W. G. Song, D. M. Marcus and J. B. Nicholas, Acc. Chem. Res., 2003, 36, 317 CrossRef CAS.
- M. Stocker, Microporous Mesoporous Mater., 1999, 29, 3 CrossRef CAS.
- D. M. McCann, D. Lesthaeghe, P. W. Kletnieks, D. R. Guenther, M. J. Hayman, V. Van Speybroeck, M. Waroquier and J. F. Haw, Angew. Chem., Int. Ed., 2008, 47, 5179 CrossRef CAS.
- M. Bjorgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195 CrossRef CAS.
- A. A. Avidan, D. F. King, T. M. Knowlton and M. Pell, Kirk–Othmer Encyclopedia of Chemical Technology, Wiley-Interscience, Hoboken, NJ, 5th edn, 2004, vol. 3 Search PubMed.
- J. M. Matsen, Powder Technol., 1996, 88, 237 CrossRef CAS.
- S. S. Chauk, L.-S. Fan, Heat Transfer in Packed and Fluidized Beds, 3rd edn, McGraw-Hill, New York, 1998 Search PubMed.
- T. M. Knowlton, S. B. R. Karri and A. Issangya, Powder Technol., 2005, 150, 72 CrossRef CAS.
- J. Diebold and J. Scahill, Prepr. Pap. - Am. Chem. Soc., Div. Fuel Chem., 1987, 32, 297 Search PubMed.
- P. A. Horne and P. T. Williams, Fuel, 1996, 75, 1043 CrossRef CAS.
- P. A. Horne, N. Nugranad and P. T. Williams, J. Anal. Appl. Pyrolysis, 1995, 34, 87 CrossRef CAS.
- Y. Tsuchiya, H. Shimogaki, H. Abe and A. Kagawa, J. Wood Sci., 2010, 56, 53 CrossRef CAS.
- P. R. Patwardhan, J. A. Satrio, R. C. Brown and B. H. Shanks, Bioresour. Technol., 2010, 101, 4646 CrossRef CAS.
- D. C. Elliott, K. L. Peterson, D. S. Muzatko, E. V. Alderson, T. R. Hart and G. G. Neuenschwander, Appl. Biochem. Biotechnol., 2004, 115, 807 CrossRef.
- Y. T. Kim, K. D. Jung and E. D. Park, Microporous Mesoporous Mater., 2010, 131, 28 CrossRef CAS.
- C. H. Ding, X. S. Wang, X. W. Guo and S. G. Zhang, Catal. Commun., 2008, 9, 487 CrossRef CAS.
- S. J. Wang, G. W. Guo, L. Q. Zhao and R. H. Wang, Chin. J. Catal., 1992, 13, 38 Search PubMed.
- G. L. Woolery, G. H. Kuehl, H. C. Timken, A. W. Chester and J. C. Vartuli, Zeolites, 1997, 19, 288 CrossRef CAS.
-
G. M. Smith and B. K. Speronello, vol. US pat, 7
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 547813 B2 (ed. USPO), BASF Catalysts LLC, United States, 2009.
547813 B2 (ed. USPO), BASF Catalysts LLC, United States, 2009. - Knocking Characteristics of Pure Hydrocarbons (Research Project 45), American Society of Testing Materials (ASTM), Special Technical Publication No. 225, Philadelphia, PA, 1958 Search PubMed.
- Clean Air Act of 1990 report of the Committee on Energy and Commerce, United States Congress House Committee on Energy and Commerce, USGPO, 1990 Search PubMed.
- T. Sano, N. Yamashita, Y. Iwami, K. Takeda and Y. Kawakami, Zeolites, 1996, 16, 258 CrossRef CAS.
- I. Wang, T. J. Chen, K. J. Chao and T. C. Tsai, J. Catal., 1979, 60, 140 CrossRef CAS.
- V. R. Choudhary and D. B. Akolekar, J. Catal., 1990, 125, 143 CrossRef.
- S. M. Campbell, D. M. Bibby, J. M. Coddington, R. F. Howe and R. H. Meinhold, J. Catal., 1996, 161, 338 CrossRef CAS.
- E. Rautiainen, R. Pimenta, M. Ludvig and C. Powels, Catal. Today, 2009, 140, 179 CrossRef CAS.
- C. A. Trujillo, U. N. Uribe, P. P. KnopsGerrits, L. A. Oviedo and P. A. Jacobs, J. Catal., 1997, 168, 1 CrossRef CAS.
- E. L. Kugler and D. P. Leta, J. Catal., 1988, 109, 387 CrossRef CAS.
- M. T. Xu, X. S. Liu and R. J. Madon, J. Catal., 2002, 207, 237 CrossRef CAS.
- J. Werther and J. Reppenhagen, AIChE J., 1999, 45, 2001 CrossRef CAS.
- C. Pereira, G. T. Kokotailo and R. J. Gorte, J. Phys. Chem., 1991, 95, 705 CrossRef CAS.
- J. H. Jae, G. A. Tompsett, Y. C. Lin, T. R. Carlson, J. C. Shen, T. Y. Zhang, B. Yang, C. E. Wyman, W. C. Conner and G. W. Huber, Energy Environ. Sci., 2010, 3, 358 RSC.
- D. J. Nowakowski, A. V. Bridgwater, D. C. Elliott, D. Meier and P. de Wild, J. Anal. Appl. Pyrolysis, 2010, 88, 53 CrossRef CAS.
- D. Mohan, C. U. Pittman and P. H. Steele, Energy Fuels, 2006, 20, 848 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |