Highly dispersed silica-supported nanocopper as an efficient heterogeneous catalyst: application in the synthesis of 1,2,3-triazoles and thioethers†
Pitchaimani
Veerakumar
a,
Murugesan
Velayudham
b,
Kuang-Lieh
Lu
b and
Seenivasan
Rajagopal
*a
aDepartment of Physical Chemistry, School of Chemistry, Madurai Kamaraj University, Madurai, 625 021, India. E-mail: rajagopalseenivasan@yahoo.com; spveerakumar@gmail.com; Fax: +91 452 2459105; Tel: +91 452 2458246
bInstitute of Chemistry, Academia Sinica, Taipei, 115, Taiwan
First published on 7th September 2011
Abstract
In this paper, we report the synthesis of amine modified SiNPs (silica nanoparticles) by a sol–gel method and the role of synthesized SiO2 as a solid support for the nanocatalyst CuNPs (copper nanoparticles). The nanocatalyst is characterized by XRD, HRTEM, BET, AFM, SEM, EDX, UV-vis, FT-IR and TGA techniques. The Cu/SiO2 (catalyst A) serves as an efficient heterogeneous nanocatalyst exhibiting high catalytic activity for the synthesis of a series of 1,4-disubstituted-1,2,3-triazoles and thioethers. The catalyst A can be recycled and reused several times without any significant loss of catalytic activity as proved by XRD and HRTEM techniques.
Introduction
In the past four decades, after the first report in 1965, much attention has been devoted to the Huisgen 1,3-dipolar cycloaddition reaction of organic azides with alkynes,1 and the term “Click chemistry”, first introduced by Sharpless and co-workers in 2001, is now widely used for this reaction.2,3 Meldal et al.4 and Sharpless et al.5 have reported Cu(I)-catalyzed union of terminal alkynes and organic azides to give 1,4-disubstituted triazoles. Cu(I)-catalyzed cycloaddition of water insoluble aliphatic/aryl azides with alkynes at room temperature has been extensively reported.6a–c Apart from the Cu complexes, Pd7 and Ru8a,b complexes have also been found to be efficient catalysts for the synthesis of 1,5-disubsituted triazoles. Cu(I)-zeolites9a–c and CuII-hydrotalcite10 serve as efficient heterogeneous catalysts for Huisgen (3 + 2) cycloaddition reactions. Microwave (MW) irradiation11 and ultrasound (US) methods12 accelerate the nanocopper catalyzed azide–alkyne cycloaddition reactions. The products of the reaction exhibit remarkably broad scope in the field of metal ion sensors,13a,b as chelating agents,14 and in medicinal chemistry.15a,bNanomaterials are of topical interest, because of their intriguing properties different from those of their corresponding bulk materials.16 Due to their unique properties, nanomaterials are employed in electronic, optical, catalytic, coating, medical and sensor applications.17 Metal nanoparticles are very attractive catalysts compared to bulk catalysts since they have a high surface to volume ratio and their surface atoms are very active.18 Numerous review articles highlight the use of many different types of organic and inorganic reactions of noble metal nanoparticles suspended in colloidal solutions as well as those adsorbed onto different supports as catalysts.19a–d Copper and copper oxide nanoparticles are of significant technological interest.20 Copper, in the nanoform, is known for the past one decade to show fascinating catalytic activity for the various organic reactions.21a–d Copper nanoparticles are of great interest in a broad technological arena including catalysis and energy conversion.22,23
Copper is less expensive compared to Au, Ag, Pd, Pt, Ru and Rh which have been extensively used as catalysts for organic transformations.24 The size of the nanocatalyst is of utmost importance in catalysis for providing a highly active catalyst surface, which maximizes the reaction rates and minimizes consumption of the catalyst.21 The tunability of size and spacing of metal nanoparticles with the polymer PEI (polyethyleneimine) opens a new way to synthesize nanomaterials with controlled diameter. This leads to the tuning of catalytic activity with the change of size of nanoparticles in the range 20–100 nm.25a–c The capping layer of the CuNPs can be varied or modified, thus providing a unique possibility to control their surface and catalytic properties. We have synthesized the low cost CuNPs with a specific size, well defined surface composition, isolable and redispersible properties.
Polyvinylpyrrolidone (PVP) and surfactant stabilized CuNPs,26a,b nanostructured CuO materials,27 Cu/C,28a,b Cu/CuO,29 hetero bimetallic Cu–Ni/C30 and Cu/AlO(OH)31 have been widely employed as catalysts for “Click” reactions. The heterogeneous Cu/SiO2 catalyst has been used as the active catalyst in hydroxylation of phenol using hydrogen peroxide32 and for the selective conversion of ethanol to acetaldehyde.33
The formation of a C–S bond represents a key step in the synthesis of many organic molecules that are of biological, pharmaceutical, and materials science interest.34a,b For example, a large variety of aryl sulfides are in use for diverse clinical applications, particularly for the treatment of cancer35 and human immunodeficiency virus diseases36 and for photoinduced electron transfer reactions.37a–d In recent years, transition metal nanoparticles catalyzed cross-coupling reaction of aryl halides with thiols has been developed into a versatile and efficient method for a variety of synthetic organic transformations.38a–d In order to obtain high catalytic activity, metal nanoparticles are generally dispersed on support materials, which offer high thermal and chemical stabilities combined with a well-developed porous structure and high surface area, meeting the requirements for most applications.39 Nanoparticles can also be easily prepared and further functionalised, adding value to their use as support or catalyst. Depending on the chemical reactivity of the support, metal oxides can be classified as inert (e.g. SiO2) and reactive (e.g. CeO2) metal oxides. Among the metal oxides, SiO2,40 Al2O3,41 TiO2,42 CeO243 and ZrO244 are the most commonly employed supports.
The goal of this work is the development of new, highly efficient catalyst systems for the cycloaddition and C–S coupling reactions using copper nanocatalysts which can be recycled and reused several times without any loss of their catalytic activity. The catalytic activities of nano Cu/SiO2 for the 1,3-diploar cycloaddition and C–S coupling reactions as detailed in Scheme 1 are investigated and presented in this article.
 | ||
| Scheme 1 Synthesis of 1,2,3-triazoles and thioether using catalyst A. | ||
Experimental
Materials and reagents
Copper(II) chloride (CuCl2), highly branched polyethyleneimine (PEI, MW ≈ 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), tetraethyl orthosilicate (TEOS, 98%), sodium azide (NaN3), 3-aminopropyl triethoxysilane (APTES, 99%), phenylacetylene, thiols, derivatives of iodo- and bromobenzene and NaBH4 (Merck) were purchased from Sigma Aldrich and used as received. Water purified through a double distilled system was used.
000), tetraethyl orthosilicate (TEOS, 98%), sodium azide (NaN3), 3-aminopropyl triethoxysilane (APTES, 99%), phenylacetylene, thiols, derivatives of iodo- and bromobenzene and NaBH4 (Merck) were purchased from Sigma Aldrich and used as received. Water purified through a double distilled system was used.
Synthesis of silica supported PEI/CuNPs
Monodisperse spherical SiNPs were synthesized by following the Stöber method45 from tetraethyl orthosilicate using liq.NH3 as catalyst. The synthesis of amino surface-modified SiNPs was carried out using the literature procedure.21b,46a,b The PEI stabilized CuNPs were prepared by the method of Pulkkinen et al.47 In a typical synthesis, 2.4 g (2 mmol) of PEI was dissolved in 150 mL of water. A sample of 269 mg (2.0 mmol) of CuCl2 was mixed with 757 mg (20.0 mmol) of NaBH4 carefully and degassed with nitrogen for about 30 min. The reaction mixture was agitated to obtain a homogeneous Cu/PEI solution under deaerated conditions. The growth of CuNPs was monitored using UV-vis absorption spectroscopy. Amine modified SiO2 (100 mg) was added to CuNPs by stirring for 30 min and sonicated for 45 min. Dark brown colored PEI/CuNPs were bound on the surface of SiNPs with elapsing of reaction time, and the reaction proceeded for 2.5 h. Finally, the particles were washed with ethanol, centrifuged and dried under vacuum. The entire synthetic procedure is given in Scheme 2. In the absence of a suitable support, metal particles aggregate, have reduced surface area and restricted control over particle size. | ||
| Scheme 2 Schematic representation of a four-step process for the synthesis of catalyst A. | ||
Synthesis of 1,2,3-triazoles
In our initial exploration, for the synthesis of 1,2,3-triazoles48 the reaction of benzyl bromide with sodium azide and phenylacetylene was chosen as a model reaction. Alkyl azides were synthesized at room temperature from the corresponding bromides by nucleophilic substitution with sodium azide in DMSO.49 First the alkyl halide and sodium azide with a 1:1.2 molar ratio were suspended on dry DMSO with vigorous stirring for 10–15 min at room temperature, 1.2 mole ratio of phenylacetylene and 0.05 mol% of catalyst A were added and stirring continued to get the corresponding 1,2,3-triazoles in good yield. The product formed was then extracted with ether and dried under reduced pressure to obtain the desired triazole as a white crystalline solid. The results are collected in Table 1.
| Entry | Azides | Alkynes | Products | Time/min | Yieldb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: azide (1.2 mmol), aryl or alkyl halide (1.0 mmol) and alkyne (1.2 mmol), catalyst A (0.05 mol%) and DMSO (5.0 mL) at RT. b Yield refers to column chromatography yield. c Alkyne 2.4 mmol used in DMSO at 15 min. d Yield after 2nd and 3rd cycles. e Silica, 3h. f 1,10-Dibromodecane (1.0 mmol), azide (2.2 mmol), and alkyne (2.4 mmol) were used. | |||||
| 1 | — |

|

|
15 | 98c |
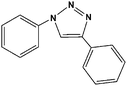
| 2 |

|

|

|
10 | 98, 92d, 89d, 0e |
| 3 |

|
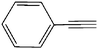
|
|
25 | 97 |
| 4 | CH3–N3 |
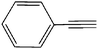
|

|
20 | 92 |
| 5 | C3H7–N3 |

|

|
35 | 83 |
| 6 | C6H13–N3 |

|

|
30 | 95 |
| 7 | C10H21–N3 |

|
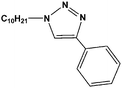
|
35 | 95 |
| 8 | C12H25–N3 |
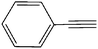
|

|
30 | 65 |
| 9 | C16H33–N3 |
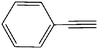
|

|
35 | 62 |
| 10 | N3–C10H20–N3 |

|

|
45 | 60f |
| 11 | — |

|

|
15 | 96 |
| 12 |

|
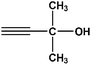
|

|
60 | 65 |
| 13 |

|

|

|
60 | 95 |
| 14 | C3H7–N3 |

|

|
55 | 90 |
| 15 | C6H13–N3 |

|

|
50 | 94 |
In addition, the great difficulty in purification of the product when the reaction is incomplete is that some alkyl azides decompose rapidly with danger of explosion or distilling.49 Furthermore, alkyl azides, generally, have boiling temperature adjacent to that of the corresponding alkyl bromides. Thus, we decided to initiate a systematic study of the versatility of the nucleophilic substitution of bromide utilizing NaN3 in DMSO at ambient temperature. Herein, we discuss the successful preparation, in high yield of various alkyl azides in excellent purity (see ESI†).
C–S coupling reaction
A stirred solution of aryl or alkyl thiol (1.0 mmol) was mixed with aryl halide (1.1 mmol), catalyst A (1.5 mol%), and KOH (1.5 mmol) in DMSO (3 mL). The solution was heated at 110 °C in a N2 atmosphere. The progress of the reaction was monitored by TLC. The reaction mixture was then cooled to room temperature and treated with diethyl ether (10 mL). The aqueous layer was separated, extracted with diethyl ether (2 × 5 mL), and dried over anhydrous Na2SO4. The combined organic extracts were concentrated in vacuum, and the resulting product was purified by column chromatography on silica gel with a mixture of ethyl acetate and n-hexane (2:8) as eluent to afford analytically pure C–S cross-coupling products. Aryl and alkyl sulfides were isolated in the yields reported in Table 2. Products were characterized using 1H NMR and 13C NMR spectra given in ESI.†
| Entry | Halides | Thiols | Products | Time/h | Yieldb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: thiol (1.2 mmol), aryl halide (1.0 mmol), catalyst A (1.5 mol%), KOH (1.5 mmol), and DMSO (3.0 mL) at 110 °C, under an N2 atmosphere. b Yields of isolated products. c Isolated yields after 2nd and 3rd cycles. d Bromo acetophenone is used. | |||||
| 1 |
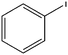
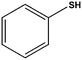
|

|

|
9 | 96, 90c, 82c |
| 2 |

|
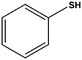
|

|
12 | 91 |
| 3 |
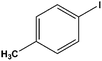
|

|

|
12 | 89 |
| 4 |
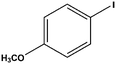
|
|

|
10 | 92 |
| 5 |
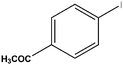
|

|

|
11 | 90 [83]d |
| 6 |
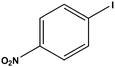
|
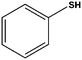
|

|
10 | 87 [75]d |
| 7 |

|
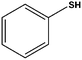
|

|
12 | 70 [65]d |
| 8 |

|

|

|
11 | 85 |
| 9 |

|

|

|
11 | 88 [80]d |
| 10 |

|
C2H5–SH |

|
11 | 91 |
| 11 |

|
C12H25–SH |
|
12 | 92 |
| 12 |

|
C12H25–SH |

|
11 | 90 [85]d |
Instrumentation
The X-ray powder diffraction (XRD) pattern of the catalyst was taken using a XPERT-PRO diffractometer operated at a voltage of 40 kV and a current of 30 mA with Cu Kα radiation (λ = 1.5406 Å). High-resolution TEM (HRTEM) images and the selected area electron diffraction (SAED) patterns were performed on a JEOL 3011 instrument with an accelerating voltage of 300 kV. The catalyst A was dispersed on ethanol solution under ultrasonic vibration for 20 min and one drop of the suspension evaporated onto a carbon-coated copper grid for HRTEM measurement. N2 adsorption–desorption isotherms of the samples at 77 K were obtained on a Micromeritics ASAP 2020 instrument. SEM observations were carried out on a JEOL JSM-6390 electron microscope. Detailed composition characterization of the Cu/SiO2 composites was carried out with energy-dispersive X-ray (EDX) analysis (equipped with the SEM). A thermogravimetry (TGA) measurement was done using NETZSCH STA 409PC equipment under a flowing nitrogen atmosphere. The temperature range was 25–700 °C. UV-visible absorption spectral measurements were carried out with a SPECORD S100 diode-array spectrophotometer. FT-IR spectra of the purified SiNPs were recorded using a 8400 S Shimadzu FT-IR spectrometer in the region 4000–400 cm−1 with a spectral resolution of 2 cm−1. Solid samples were dispersed in dry KBr discs at room temperature. Atomic force microscopy (AFM) (APE Research nanotechnology, AFM A100 SGS), working at 100 kV was used to measure the size of these nanoparticles. The samples were prepared by evaporating a drop of the dilute aliquot solution onto a thin glass plate. 1H and 13C NMR data of products were acquired on a Bruker 300 MHz NMR spectrometer with CDCl3 as the solvent.Results and discussion
Unsupported or in the absence of surface capping agents and stabilizers, the surface atoms of metallic nanoparticles with their typically high surface energy become susceptible to aggregation into bulk material. To overcome this problem, catalytic nanoparticles are immobilized on solid supports.19a–d Surface coverage by a polymer is advantageous because in addition to stabilizing and protecting the nanoparticles, polymers offer unique possibilities for modifying both the environment around catalytic sites and access to these sites. Hence the protective polymer not only influences particle size and morphology but it is likely to have a tremendous influence on catalytic activity and selectivity.23 According to Scheme 2 we have used the four step process to synthesize the PEI/Cu NPs supported onto the SiO2 surface: (i) synthesis of the SiNPs, (ii) amine functionalization of the SiO2 surface, (iii) synthesis of the PEI/CuNPs, and (iv) attachment of the PEI/CuNPs onto the amine functionalized SiO2 surface. In the presence of the reducing agent, the Cu2+ ion is reduced to Cu0 on the polymer (Scheme 2), preventing the agglomeration of the metallic nanoparticles. PEI/CuNPs are covalently attached to the functionalized SiO2 colloids via the Cu–N bond. The silica colloids are functionalized with APTES which is confirmed by FT-IR spectroscopy. In this paper, we examine the surface properties of a silica nanosphere and demonstrate the presence of surface silanol groups (–SiOH) which can be used to sequester active Cu sites for the selective formation of 1,2,3-triazoles and thioethers.XRD studies
X-Ray powder diffraction (XRD) data (Fig. 1) are used for the identification and quantification of the crystalline phases, along with the measurement of the crystallite size. XRD peaks for SiO2 were not observed and thus could be attributed to the amorphous nature. XRD analysis of the catalyst A revealed its metallic nature (Fig. 1). All the samples exhibited face centered cubic (fcc) structure. | ||
| Fig. 1 XRD pattern of catalyst A. | ||
XRD patterns are analyzed to determine peak intensity, position and width. Full width at half-maximum (FWHM) data are used with the Scherrer's formula to determine the mean particle size.50 All Bragg's reflections at 2θ = 43.5, 50.6, and 74.3 can be indexed as the [111], [200] and [220] planes of copper. It obviously indicates that CuNPs are in the Cu(0) state, not as oxidized species (CuO, Cu2O) and no impurity diffraction peaks are detected.51a–c After the reaction is complete, the recovered catalyst A is checked with its XRD analysis and the details are given in the (ESI†, S6). This is to be expected for small CuNPs and more so for those embedded within the silica shell. Nevertheless, the diffraction patterns clearly indicate the presence of Cu and no significant copper oxide phase.52
HRTEM analysis
HRTEM images of monodisperse spheres of bare SiNPs with a smooth surface and a homogeneous size are shown in Fig. 2. All the particles are spherical and the diameter of the particles depends on the preparation conditions (TEOS concentration and reaction time). | ||
| Fig. 2 HRTEM images of (a) silica nanoparticles and (b) amino modified SiNPs. | ||
The measured average size of the bare SiO2 spheres obtained without addition of CuNPs is 180 ± 30 nm. Fig. 3 shows the HRTEM images of catalyst A nanospheres prepared by using the modified Stöber method and the size of CuNPs is about 5 ± 2 nm. The HRTEM measurements show that SiNPs are homogeneously attached with CuNPs (Fig. 3a). The inset in Fig. 3d shows a typical selected-area electron diffraction pattern of the CuNPs which reveals the characteristic (111), (200) and (220) diffraction peaks of metallic copper, indicating the formation of a crystallized state in the face centered cubic (fcc) structure in accordance with the JCPDS file no 04-836.53
 | ||
| Fig. 3 HRTEM photographs of catalyst A. Here (a), (b) and (d) correspond to the different images of the representative catalyst A; (c) shows EDX analysis of catalyst A and inset (d) shows SEAD spectrum of catalyst A. | ||
After the reaction is complete, the recovered catalyst is checked with its HRTEM image given in the ESI† (S7). Interestingly it is observed that the shape and size of the particles remain unchanged and support the proposal that the morphology of the catalyst remains the same even in the used conditions. Analysis through energy dispersive X-ray (EDX) spectrometers confirmed the presence of elemental copper and silicon signals from the catalyst A (Fig. 3c). The vertical axis displays the number of X-ray counts whilst the horizontal axis displays energy in keV. Identification lines for the major emission energies of Si metal from the catalyst A are displayed and these correspond with peaks in the spectrum, thus giving confidence that copper has been correctly identified. These results support our conclusion that SiO2 particles construct the surface layer of the CuNPs.
BET surface area studies
N2 adsorption–desorption isotherms of the samples at 77 K are obtained on a Micromeritics ASAP 2020 instrument. Before N2 adsorption, catalyst samples are evacuated for 4 h at 350 °C. The Brunauer–Emmett–Teller (BET) equation is used to calculate the surface areas, pore volumes and pore sizes estimated at a relative pressure of 0.99.The Barrett–Joyner–Halenda (BJH) method applied to the desorption isotherm is used to determine the pore diameter distribution.54 The surface area of the synthesized silica is found to be 115.9 m2 g−1, when the CuNPs are loaded on the surface of pure SiO2. The BET surface area and the pore volume decrease from 115.9 to 104.41 m2 g−1 and 0.60 to 0.22 cm3 g−1 respectively. The N2 sorption isotherms of the calcined pure SiO2 and Cu/SiO2 catalysts are presented in ESI† (S8). It can be seen that the samples of pure SiO2 and Cu/SiO2 exhibit Langmuir type IV isotherms.54 A sharp inflection of the adsorption and desorption isotherms, in particular around P/Po = 0.9728 (for N2 at 77 K), indicates a forced closure of the hysteresis loop. In ESI† (S8a) the inset figure shows a sharp inflection of the adsorption and desorption isotherms in the P/Po range 0.8–1.0 (dotted square) emphasizing the TSE (tensile strength effect) at P/Po = 0.97.The (4V/A) term used in the estimation of pore average sizes corresponds to the assumed cylindrical model of pores. However, this assumption of cylindrical model of pores is also cited in BJH estimates of pore volume and surface area distributions. The catalyst A shows that the adsorption and desorption isotherm value at P/Po = 0.9730. According to these measurements, the BET surface area, Langmuir surface area, pore volume and pore diameter are calculated and the data are given in Table 3.
| Sample | Cu content/wt% | BET surface area/m2 g−1 | Langmuir surface area/m2 g−1 | t-Plot external surface area/m2 g−1 | Average pore diameter/nm | Pore volume/cm3 g−1 |
|---|---|---|---|---|---|---|
| SiO2 | — | 110.8 | 186.9 | 126.9 | 33.0 | 0.60 |
| Catalyst A | 1.5 | 104.1 | 168.6 | 115.5 | 27.6 | 0.21 |
SEM analysis
The synthesized Stöber method45 SiNPs are analyzed by using scanning electron microscopy (SEM). The silica particles show very uniform spherical morphology and monodisperse distribution. The mean diameter of these particles is ∼180 nm, with very little distribution in particle size, and representative electron micrographs obtained are presented in the ESI† (S9).AFM analysis
The AFM method is used to provide an independent and quantitative measurement of catalyst A particle size in the sol. The objective is to image the individual particles of catalyst A on a flat substrate using the tapping mode (contact mode) AFM technique. The size and morphology of the catalyst A are clearly seen from the two-dimensional (2-D) and three-dimensional (3-D) AFM images given in the ESI† (S9). By achieving a good dispersion of the nanoparticles it is possible to image them individually using AFM. This technique enables measuring the size of a particle, considering the particle's height rather than its width because the particle may be distorted by the AFM tip geometry. Individual Cu/SiO2 particles are clearly visible in this image. The diameter of each particle is estimated by examining the height profile or bearing ratio mapped by the AFM tip. Spherical particles are assumed, and the height measured by AFM is taken as the particle diameter. The average size of the copper nanostructures is estimated to be 5–6 nm according to the voltage profile of AFM images.UV-vis absorption spectral analysis
Fig. 4 shows the slow reduction of copper ions leading to the formation of CuNPs which is confirmed by SPR (Surface Plasma Resonance) peak that appeared at 583 nm.55 The CuNPs supported on SiNPs are extremely stable for several months in a N2 atmosphere. There is substantial colour change during the reduction of copper salt and the details are shown in Fig. 4. | ||
| Fig. 4 UV-visible absorption spectra of formation of PEI/CuNPs: (a) Cu2+solution (b) mixture of Cu2+ and PEI solution and (c) after the addition of NaBH4. | ||
FT-IR spectral analysis
FT-IR spectra of pure SiO2, amino modified SiO2 and catalyst A are recorded in the regions 2700–3500 and 1300–2000 cm−1. The FT-IR spectral assignments of dried and calcined samples of pure SiO2, amino modified SiO2 (NH2-SiO2) and copper coated SiO2 (catalyst A) are shown in Fig. 5 and the relevant data collected in the ESI† (S10). The 2929 and 2856 cm−1 bands are assigned to νCH of the –CH2 groups, and the bands at 1544 cm−1 and 1542 cm−1 to δNH of the –NH2 groups, both of which are associated with amino modified SiO2 and catalyst A skeleton respectively.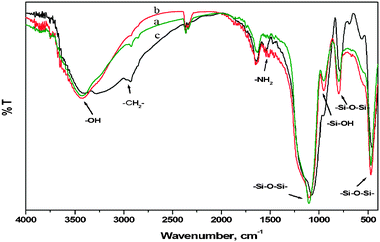 | ||
| Fig. 5 FT-IR spectra of (a) pure SiO2, (b) pure NH2-SiO2 and (c) catalyst A. | ||
The strong peaks at 1076 and 790 cm−1 are due, respectively, to νasym(Si–O) and νsym(Si–O) of the Cu/SiO2 skeleton.56a–c Moreover, the presence of the amino groups confirms the formation of the amine-modified particles. The broad peak centered at 3400 cm−1 is an envelope of νO–H for the adsorbed water, silanol groups, and νN–H of the amino groups. It should be pointed out here that the band corresponding to the Si–OH group appears at 960 cm−1 as a shoulder (arrow) of the 1076 cm−1 Si–O–Si skeleton peak, and is little weaker than that found ever in the conventional SiO2 particles that exhibit a clear and well-defined peak of the Si–OH groups.
TGA analysis
The synthesized catalyst A is initially examined by thermogravimetric analysis (TGA) under a nitrogen atmosphere in the temperature range between room temperature to 700 °C. The weight loss of Cu/SiO2 bare silica below 200 °C is 7.5% which is attributed to the physisorbed water and residual organic solvent. Identification of the evolved gas fragments or decomposition components confirms the presence of the spacer 3-aminopropylsilane (APTES) on the synthesized catalyst.28b The thermogravimetric profile of the catalyst A shows removal of a small amount of water and APTES as indicated in the two-step degradation process given in the ESI† (S11).The high temperature required to decompose and evaporate the organic content of the modified silica particles demonstrates that the silane-coupling agent is strongly bound to the particle surface and this is likely through a covalent bond.26c As for Cu/SiO2, the beginning of decomposition at 300 °C corresponds to the decomposed temperature of PEI covalently attached to nano silica particles. The Cu/SiO2 nanoparticles (ESI†, S11) show a decomposition peak with a maximum rate of decomposition at 150 °C and 8.11% of weight loss at 700 °C, attributed to the possible progress in sol–gel reaction and to the decomposition of the surface organic phase. This weight loss can be attributed to the thermal decomposition of a little amount of PEI and APTES to other decomposable organic materials present in the modified silica nanoparticles. Pulkkinen et al.47 have reported the TGA data which also show that the CuNPs bound to a trace amount of a protecting agent (PEI) are decomposed at lower temperature than the free PEI.
Audebert and Pourabbas et al.26d,e have estimated the weight loss of polypyrrole (PPy) on the surface of SiNPs as approximately 10 wt% using the TGA method. A similar method is used by us to calculate decomposition peak data of catalyst A. The entire products contain ∼12–15 wt% of volatile components, regardless of the reaction time. This indicates that longer synthesis time will not increase the amount of protecting agent left on the particle.
Optimization of click reaction
To optimize the reaction conditions, benzyl bromide, phenyl acetylene and NaN3 are selected as the test reagents for the synthesis of 1-benzyl-4-phenyl-1,2,3-traizole to examine the effect of catalyst A (0.05 mol%), at room temperature. The results are evaluated qualitatively using TLC (Table 1). To get the maximum yield the best condition is that the reagents, catalyst A, phenyl acetylene, benzyl bromide and NaN3, have to be taken in the ratio 0.05:1.2:1.0:1.2 mmol at room temperature. The reaction is allowed to continue for 10 min using DMSO as solvent. Synthetic procedure for the synthesis of azides and their corresponding NMR spectra of products are given in the ESI† (S19 and S20).
An increase in the amount of catalyst A from 0.05 to 2.0 mol% has negligible effect on the efficiency of the reaction. Using these optimized conditions, the reaction of various terminal acetylenes (phenyl acetylene, 2-methylbut-3-yn-2-ol), benzyl/alkyl halides and NaN3 is investigated (Scheme 1). It is found that all the reactions proceed smoothly to give the corresponding 1,4-disubstituted-1,2,3-triazoles in high yield, 98% (Table 1, entry 2), which clearly indicates the generality and scope of the reaction with respect to terminal alkynes and halides. When phenyl acetylene (2.4 mmol) alone is added to catalyst A (0.05 mol%) it affords 1,4-diphenylbuta-1,3-diyne and 2,7-dimethylocta-3,5-diyne-2,7-diol as homocoupling products (Table 1, entries 1 and 11). Thus, this procedure can also be utilized for homocoupling reactions. The 1H and 13C NMR spectra of homocoupling products are collected and given in the ESI† (S21 and S22).
Influence of different solvents on catalyst A catalyzed click reaction
After stabilizing the optimum conditions for the reaction we turn our attention to investigate the effect of changing the solvent on the efficiency of the coupling reaction (see the ESI†, S14). When the reaction is conducted in water, ethanol and dimethyl sulfoxide the yield of the products is good to excellent (85, 90 and 98%, respectively; ESI†, S14, entries 2, 5 and 6). The use of toluene, acetonitrile, dichloromethane and dioxane as solvents leads to the lower yields of the products (ESI†, S14, entries 1, 3, 4 and 7). During our optimization studies, various solvents are examined and it is found that the solvent plays a significant role in terms of reaction rate, isolated yield, and selectivity. The reaction of benzyl bromide with phenylacetylene and sodium azide in the presence of 0.05 mol% of catalyst A in DMSO furnished the 1,4-disubstituted triazole product in 98% yield after stirring for 10 min at ambient temperature (Scheme 1).The use of DMSO as the suitable solvent in this study deserves comments. Generally, benzyl bromide and phenylacetylene have poor solubility in water; it required more than 75 min for the completion of the reaction in water at ambient temperature. However, our interest is to develop a more efficient system having high reactivity within a short reaction time to give good yields. The use of DMSO in Click coupling reactions results in an increase in the activity of catalyst A and the yield is 98%. In terms of the sufficient solubility of azide, alkyl/aryl halides and alkynes also DMSO is a suitable medium. In order to show the efficiency of the DMSO solvent system, the same reactions were also performed in EtOH (ESI†, S14, entries 2 and 6) but it afforded 90% yield after 25 min. Generally DMSO is a relatively inexpensive, stable and environmentally compatible solvent in organic and inorganic synthesis due to its specific chemical and physical properties.57 The advantage of DMSO as the solvent is attributed to: (i) it exerts sufficient interaction with the surface of the metal nanoparticles to effectively passivate and stabilize the nanoparticle dispersion as created within this solvent medium. (ii) DMSO acts as a stabilizer during the reaction, no precipitation/agglomeration occurs (see in ESI†, S7). (iii) Cu does not suffer from aerobic oxidation during the reaction at ambient temperature (DMSO is acting as an additional capping agent in this case). The points (i)–(iii) suggest that DMSO is a sound reaction medium/solvent for the synthesis of 1,2,3-triazoles, that meets contemporary demands for more benign conditions. The other solvents do not possess this type of vital properties.
Comparison with other catalyst systems
In order to evaluate the efficiency of the different supporting material carrying copper nanocatalyst, we have compiled the data on the synthesis of 1,2,3-triazoles using copper catalysts and the data collected in Table 4. The data in Table 4 show that although the yields are comparable for many catalyst systems, and mol% and the size of CuNPs used in the present study are smallest and the selectivity is better than the other systems i.e., metal nanoparticles are very attractive catalysts compared to bulk catalytic materials due to their high surface-to-volume ratio. The data in Table 4 evidently indicate that the catalyst A is smaller in size compared to alumina supported CuNPs,48 PVP capped CuNPs,26a Cu(0) nanosized activated powder,58 CuNPs in ionic liquids59 and Cu/C28b catalyst system. The novelty of this catalyst system is that a lesser quantity of catalyst (0.05 mol%) is needed compared to other catalyst systems reported.| Entry | Catalysta (%) | Size/nm | Mol (%) | Time/h | Solvent | Yield (%) |
|---|---|---|---|---|---|---|
| a References. b 65 and 100 °C temperatures were maintained. c Reaction conditions as exemplified in the experimental procedure. Here L = Ligand, IL = Ionic Liquid. | ||||||
| 1 | PVP-Cu26a | 10–35 | 5.0 | 20 min | Formamide | 91 |
| 2 | Cu nanoclusters26b | 1.6–2.1 | 0.01 mmol | 18 | H2O/t-BuOH | 80 |
| 3 | CuO27 | >20 | 5 | 3 | H2O/t-BuOH | 98 |
| 4 | Cu/C28b | 80–300 | 1 | 0.6 | H2Ob | 91 |
| 5 | Cu/CuO29 | 14 | 13–20 | 3–4 | Toluene | 95 |
| 6 | Cu/AlO(OH)31 | 5–8 | 6 | 6 | n-Hexane | 94 |
| 7 | Cu/Al2O348 | >100 | 10 | 3–8 | H2O | 92 |
| 8 | Cu nanopowder58 | 50–60 | 10 | 2 | H2O/t-BuOH | 90 |
| 9 | CuNPs/IL59 | 80–130 | 5.0 | 18 min | — | 89 |
| 10 | CuNPs/L60 | 3.0 ± 1.5 | 10 | 10 | THFb | 98 |
| 11 | Pure SiO2 | >100 | 0.05 | 3 | DMSO | Tracec |
| 12 | NH2–SiO2 | >100 | 0.05 | 3 | DMSO | 25c |
| 13 | Catalyst A | 5 ± 2 | 0.05 | 10 | DMSO | 98c |
| 14 | CuCl2 | — | 0.05 | 3 | DMSO | 0c |
| 15 | — | — | — | 10 | DMSO | 0c |
The additional advantage with the present system is that the reaction is conducted at room temperature but in the unsupported CuNPs28c,60 and Cu/C27 the reaction conditions 65 and 100 °C were maintained. In order to find the role of Cu/SiO2 in the Huisgen 1,3-dipolar cycloaddition reaction, we have carried out the reaction in the presence of silica (SiO2), amino modified silica (SiO2–NH2), and CuCl2 under similar experimental conditions but we got lower yields and the results are summarized in Table 4. Thus, it is concluded that the heterogeneous catalyst A catalyzes the reaction efficiently and the corresponding triazole is obtained in high yield with a low amount of catalyst.
The role of PEI as a stabilizer
PEI is a hydrophilic polymer with primary (25%), secondary (50%) and tertiary (25%) amino groups and carries an overall positive charge in the neutral aqueous solution. Because of its abundant positive charge, it is widely used as the stabilizer for the CuNPs to achieve surface functionalization.61 It also stabilizes Ag, Au, and Pd metal nanoparticles62a–c and semiconductor quantum dots (QDs).63 When NaBH4 is added to CuCl2 and PEI mixture, colloidal nucleation is achieved and the nanoparticles begin to grow. Under vigorous stirring, PEI diffused quickly to the colloids and is adsorbed on the surface of colloids because of the electrostatic interaction between positively charged PEI and small negatively charged CuNPs which results in the positively charged colloids. The high cationic charge density and the “steric” effect of PEI kept each nanoparticle apart which results in the formation of nanoparticles stable up to three months without any agglomerization.Proposed mechanism for click reaction
A reaction mechanism proposed for the CuNPs catalysed 1,3-dipolar cycloaddition of terminal alkynes with azides is outlined in Scheme 3 on the basis of the previous reports.28b,c | ||
| Scheme 3 Reaction mechanism proposed for the catalyst A catalyzed Click reaction. | ||
The proposed mechanism (Scheme 3) for the reaction is similar to the one established in an earlier report.6c During the reaction, it is proposed that CuNPs are attracted towards the phenylacetylene to form a Cu(I)–acetylidine complex in step I (Scheme 3). Formation of the Cu(I)–acetylidine complex by initial coordination between CuNPs and alkyne is followed by the addition to azide group to give 1,2,3-triazole. The proposed mechanism involves the following steps: (1) conversion of the alkyne to the Cu–acetylidine, (2) addition of synthesized aryl or alkyl azides for attachment to the Cu–acetylidine, (3) formation of π-complex as an intermediate product, (4) attack of the distal nitrogen of the azide to the C-2 carbon of the Cu–acetylidine to give a six-membered metallacycle, (5) ring contraction to afford a Cu(I)–triazolide complex and (6) formation of triazole as a product. The copper in the nanoparticle state exhibits both zero as well as one oxidation state during the reaction because of its unsatisfied surface valences.21aOrgueira et al.58 have reported the use of Cu(0) nanosize activated powder as catalyst for cycloaddition between terminal alkynes and azides. During the course of the reaction, zero-valent copper gets oxidized to the Cu(II) state via the Cu(I) state, which precludes the use of catalyst for further use. For the completion of the reaction 10–15 min are required to get the product in excellent yields.
C–S coupling reaction using catalyst A
The catalyst A is also applied to C–S bond formation via coupling of thiols with aryl halides. The results show that this heterogeneous catalyst also could successfully promote the C–S coupling reactions, and the desired products are obtained in good to excellent yields when the 1.5% mole of catalyst A is used as shown in the ESI† (S15). On the other hand a slight to large decrease in yield is noticed when the amount of catalyst A is lowered from 1.5 mol% to 1.0 mol% and 0.5 mol% respectively. As a summary of the above results, the optimized system presented here involves the reaction between iodobenzene and thiophenol under DMSO neat solvent conditions at 110 °C, in the presence of 1.5 mmol of KOH and 1.5–2.0 mol% catalyst A.To check the scope of the procedure, the reaction of different thiols with three aryl halides is then studied (Table 2). On comparing the reactivity and yields of products, the iodobenzene is more reactive than bromo- and chlorobenzenes with aryl thiols (Table 2). To determine the scope of the catalytic system, the present protocol is applied to reactions of a range of commercially available aryl iodides and thiophenols. As shown in Table 2, the coupling of thiophenol with different aryl halide moieties is successful, leading to the desired products in good yields. These reaction conditions are also suitable for the coupling of different aryl/alkyl thiols with iodobenzenes (Table 2); ethane-, dodecane, and cyclohexanethiol afforded the desired cross-coupled products in 91–97% yield (Table 2, entries 8–12). The reaction of substrates with a longer alkyl chain (dodecanethiol) and of benzenethiol required slightly longer time to reach completion (Table 2, entries 1 and 11) respectively. The yields are low compared to bromo acetophenone (Table 2, entries 5, 9 and 12) and similar for the nitro and hydroxyl derivatives (Table 2, entries 6 and 7). Bromobenzene is less reactive than iodo derivatives. The coupling also proceeded well with substituted thiophenols and alkanethiols. This reaction is also very chemoselective and high yielding. In the absence of catalyst A the coupling reaction is not initiated at all. It is found that 1.5 mol% of catalyst A provides the best results in terms of reaction time and yield. DMSO is found to be the solvent of choice furnishing best results among other solvents such as EtOH and DMF.
Effect of solvents and bases on C–S coupling reaction
For this C–S coupling reaction between iodobenzene and benzenethiol different parameters were optimized to develop the scope of this reaction further. First we tried the significant dependence of the S-arylation on the nature of the different solvents and bases (ESI†, S16 and S17). The solvents EtOH and DMF are less effective than DMSO. A variety of bases were tested in which NaOH, pyridine and K2CO3 provided the arylated products in moderate to excellent yields (ESI†, S17, entries 2, 5 and 6). Other bases such as NaOAc and Na2CO3 gave trace or small amounts of diaryl sulfide (ESI†, S17, entries 1 and 3). But in the presence of KOH the DMF provides 60% yield compared to DMSO (ESI†, S16).C–S coupling of thiophenol with different aryl halides
First, we have studied the cross coupling of iodobenzene with thiophenol as the model reaction (ESI†, S18). The reaction affords the desired C–S cross-coupling product to form diphenyl sulfide in 96% yield when the substrate is stirred in DMSO at 110 °C for 9 h in the presence of 1.5 mol% of the Cu/SiO2 nanocatalyst. Finally, the influence of the amount of catalyst A is evaluated. Increasing the amount of catalyst A to 2.0 mol% has little effect on the efficiency of the coupling reaction as shown in ESI†, S18).Mechanism for C–S cross coupling reaction
Generally thiols (–SH) interact strongly with the transition metal nanoparticles. The experimental details clearly suggest that the reaction involves a heterogeneous process and the catalysis may occur on the surface of the CuNPs. Both the aryl halides and thiols adsorbed on the surface of the CuNPs couple efficiently to form the thioethers. The details of the mechanism of the reaction are shown in Scheme 4. | ||
| Scheme 4 Proposed mechanism for catalyst A catalyzed C–S coupling reactions. | ||
Evaluation of the efficiency of different catalysts on C–S coupling reaction
The results from the study on the effect of various supported nanocatalysts on the aryl sulfide formation with different conditions are shown in Table 5. Among these metal nanoparticles catalyst A is found to be a very effective catalyst for C–S coupling reaction. However, In2O3,38c CuO38a,64a,b and CuO/SiO265 nanoparticles have been reported to promote the C–S arylation without the assistance of any additional ligand. But the drawback of these systems is that more time is required for the reaction. The size and mol% of the other catalyst systems collected in Table 5 (entries 3–5 and 8) show that large size and high mol% are used in other systems.| Entry | Catalysta | Size/nm | Mol (%) | Time/h | Temp/°C | Solvent | Yield (%) |
|---|---|---|---|---|---|---|---|
| a References. b Reaction conditions as exemplified in the experimental procedure. MW-HMS = Microwave assisted-hexagonal microporous silica. IL = Ionic liquid; NP = Nanopowder. | |||||||
| 1 | In2O3NPs38c | 15–25 | 3.0 | 24 | 135 | DMSO | 97 |
| 2 | CuONPs38a | 33 | 1.26 | 10 | 80 | DMSO | 95 |
| 3 | CuONPs64a | >20 | 2.5 | 20 | 80 | DMSO | 90 |
| 4 | CuONPs64b | NP | 10.0 | 3 | 110 | IL | 99 |
| 5 | CuO/SiO265 | >50 | 5.0 | 21 | 110 | DMSO | 80–85 |
| 6 | Cu–MW–HMS66 | 2–3 | 0.05 | 10 | <100 | CH3CN | >99 |
| 7 | CuNPs38b | 4–6 | 20.0 | 5 | 120 | DMF | 98 |
| 8 | Cu/Al2O338d | >100 | 5.0 | 7 | 110 | DMF | 70–98 |
| 9 | Pure SiO2 | >100 | 1.5 | 9 | 110 | DMSO | 0b |
| 10 | NH2–SiO2 | >100 | 1.5 | 9 | 110 | DMSO | 0b |
| 11 | Catalyst A | 5 ± 2 | 1.5 | 9 | 110 | DMSO | 96b |
| 12 | CuCl2 | — | 1.5 | 9 | 110 | DMSO | 0b |
| 13 | — | — | — | 9 | 110 | DMSO | 0b |
For hexagonal microporous silica (HMS) and Cu/Al2O3 systems CH3CN and DMF are used as the solvent.38d,66 We compare the activities of SiO2, NH2–SiO2, and commercially available CuCl2 catalysts for the C–S arylation at 110 °C. All these catalysts are inactive (Table 5, entries 9, 10 and 12). The coupling reaction does not occur in the absence of the catalyst (Table 5, entry 13).
Among many supported NPs, silica-based CuNPs have been well studied because of the following reasons: (i) SiNPs are easy to synthesise at room temperature, (ii) SiNPs size can be easily tuned, (iii) easy adjustment of synthesis parameters leads to CuNPs with narrow size distribution (‘monodispersed CuNPs’), (iv) SiNPs are stable in organic solvents, and (v) they are environmentally friendly materials. Due to these attractive features, Cu/SiO2 nanoparticles found wide-spread applications in the synthesis of 1,2,3-triazole and C–S coupling reactions.
The stabilization of CuNPs with polyelectrolytes (PEI) is due to the combination of both steric and electrostatic stabilization resulting in electrosteric stabilization. Due to its electrosteric stabilization PEI capped CuNPs are beneficial and stable for long time. More importantly, the surface modification of metal nanoparticles by introducing an electrostatic environment is highly useful, for example, in a variety of catalytic applications.67 The advantage of our catalytic system over the published literature is that it exhibits good catalytic activity for the synthesis of 1,2,3-triazole and C–S coupling reaction without the assistance of any ligands.
To the best of our knowledge, we are not aware of any report demonstrating the use of Cu/SiO2 in such C–S coupling reaction. In addition, this novel catalyst A catalyzed C–S coupling protocol is of potential industrial significance because of its high yields, simplicity in operation, scaling up to multigram quantities, and environmental (no leaching of catalyst) and economical advantages using commercially available DMSO and an inexpensive, stable and recyclable heterogeneous catalyst.
Conclusions
In conclusion, we have developed a simple and reproducible synthetic method using a sol–gel technique for the preparation of highly dispersed SiO2 supported CuNPs as a recyclable heterogeneous nanocatalyst (catalyst A). The catalyst is employed for the synthesis of 1,4-disubstituted-1,2,3-triazoles via Huisgen 1,3-dipolar cycloaddition and C–S coupling reactions using DMSO as the solvent. The surface morphology, particle size and optical properties of catalyst A are determined by using XRD, HRTEM, BET, EDX, SEM, AFM, UV-vis, FT-IR and TGA spectral techniques. Furthermore, the catalyst can be recovered by centrifugation, washed with ethanol three or four times, then dried in vacuum and reused as the catalyst in the subsequent runs. The characterization of final products is done by 1H and 13C-NMR analysis. Such a wide scope demonstrated by a single catalyst has not been reported earlier and this procedure provides a convenient route to a variety of substituted 1,2,3-triazoles and organic sulfides. Further investigations on other useful applications of this catalyst are in progress. Moreover, these heterogeneous nanocatalysts are stable showing negligible Cu leaching and aggregation, and can be recycled multiple times without loss of catalytic activity.Acknowledgements
The authors thank UGC for a SRF position under Meritorious Fellowship Scheme, DST—New Delhi, for the financial support and for assistance under the IRHPA program for the NMR facility.Notes and references
- R. Huisgen, G. Szeimies and L. Moebius, Chem. Ber., 1965, 98, 4014 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- (a) F. Wang, H. Fu, Y. Jiang and Y. Zhao, Green Chem., 2008, 10, 452 RSC; (b) C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS; (c) F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210 CrossRef CAS; (d) S. Dıez-Gonzalez, Catal. Sci. Technol., 2011, 1, 166 RSC.
- J. Li, D. Wang, Y. Zhang, J. Li and B. Chen, Org. Lett., 2009, 11, 3024 CrossRef CAS.
- (a) L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, J. Am. Chem. Soc., 2005, 127, 15998 CrossRef CAS; (b) M. M. Majireck and S. M. Weinreb, J. Org. Chem., 2006, 71, 8680 CrossRef CAS.
- (a) S. Chassaing, A. S. S. Sido, A. Alix, M. Kumarraja, P. Pale and J. Sommer, Chem.–Eur. J., 2008, 14, 6713 CrossRef CAS; (b) P. Kuhn, A. Alix, M. Kumarraja, B. Louis, P. Pale and J. Sommer, Eur. J. Org. Chem., 2009, 423 CrossRef CAS; (c) T. Boningari, A. Olmos, B. M. Reddy, J. Sommer and P. Pale, Eur. J. Org. Chem., 2010, 6338 CrossRef CAS.
- K. Namitharan, M. Kumarraja and K. Pitchumani, Chem.–Eur. J., 2009, 15, 2755 CrossRef CAS.
- P. Appukkuttan, W. Dehaen, V. V. Fokin and E. V. Eycken, Org. Lett., 2004, 6, 4223 CrossRef CAS.
- G. Cravotto, V. V. Fokin, D. Garella, A. Binello, L. Boffa and A. Barge, J. Comb. Chem., 2010, 12, 13 CrossRef CAS.
- (a) Y. Yao, D. Tian and H. Li, ACS Appl. Mater. Interfaces, 2010, 2, 684 CrossRef CAS; (b) H. Li, Y. Yao, C. Han and J. Zhan, Chem. Commun., 2009, 4812 RSC.
- H. Struthers, T. L. Mindt and R. Schibli, Dalton Trans., 2010, 39, 675 RSC.
- (a) M. Whiting, J. C. Tripp, Y.-C. Lin, W. Lindstrom, A. J. Olson, J. H. Elder, K. Barry Sharpless and V. V. Fokin, J. Med. Chem., 2006, 49, 7697 CrossRef CAS; (b) D. Minond, S. A Saldanha, P. Subramaniam, M. Spaargaren, T. Spicer, J. R. Fotsing, T. Weide, V. V. Fokin, K. B. Sharpless, M. Galleni, C. Bebrone, P. Lassaux and P. Hodder, Bioorg. Med. Chem., 2009, 17, 5027 CrossRef CAS.
- (a) C. N. R. Rao, A. Muller and A. K. Cheetham, Nanomaterials Chemistry: Recent Developments and New Directions, Wiley-VCH, 2007, ISBN: 3527316647 Search PubMed; (b) C. Burda, Z. Chen, R. Narayanan and M. A. El-sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS; (c) S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064 CrossRef CAS; (d) M. Haruta, Chem. Rec., 2003, 3, 75 CrossRef CAS.
- K. J. Klabunde, Nanoscale Materials in Chemistry, Wiley-Interscience, New York, 2001 Search PubMed.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 12663 CrossRef CAS.
- (a) B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893 CrossRef CAS; (b) J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18 CrossRef CAS; (c) A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS; (d) J. D. Aiken III and R. G. Finke, J. Mol. Catal. A: Chem., 1999, 145, 1 CrossRef.
- (a) M. Samim, N. K. Kaushik and A. Maitra, Bull. Mater. Sci., 2007, 30, 535 CrossRef CAS; (b) B. Sreedhar, P. Radhika, B. Neelima and N. Hebalkar, Chem.–Asian J., 2008, 3, 1163 CrossRef CAS; (c) F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Eur. J. Org. Chem., 2010, 1875 CrossRef CAS; (d) A. Saha and B. Ranu, J. Org. Chem., 2008, 73, 6867 CrossRef CAS.
- (a) J. A. Eastman, S. U. S. Choi, S. Li, W. Yu and L. J. Thompson, Appl. Phys. Lett., 2001, 78, 718 CrossRef CAS; (b) M Kidwai, N. K. Mishra, S. Bhardwaj, A. Jahan, A. Kumar and S. Mozumdar, ChemCatChem, 2010, 2, 1312 CrossRef CAS.
- L. Ding, A. Tselev, J. Y. Wang, D. N. Yuan, H. B. Chu, T. P. McNicholas, Y. Li and J. Liu, Nano Lett., 2009, 9, 800 CrossRef CAS.
- S. W. Boettcher, J. M. Spurgeon, M. C. Putnam, E. L. Warren, D. B. Turner-Evans, M. D. Kelzenberg, J. R. Maiolo, H. A. Atwater and N. S. Lewis, Science, 2010, 327, 185 CrossRef CAS.
- (a) S. Jeong, K. Woo, D. Kim, S. Lim, J. S. Kim, H. Shin, Y. Xia and J. Moon, Adv. Funct. Mater., 2008, 18, 679 CrossRef CAS; (b) N. N. Hoover, B. J. Auten and B. D. Chandler, J. Phys. Chem. B, 2006, 110, 8606 CrossRef CAS.
- (a) S. Bhattacharjee, D. M. Dotzauer and M. L. Bruening, J. Am. Chem. Soc., 2009, 131, 3601 CrossRef CAS; (b) S. Kidambi, J. Dai, J. Li and M. L. Bruening, J. Am. Chem. Soc., 2004, 126, 2658 CrossRef CAS; (c) R. Abu-Reziq, D. Wang, M. Post and H. Alper, Chem. Mater., 2008, 20, 2544 CrossRef CAS.
- (a) A. Sarkar, T. Mukherjee and S. Kapoor, J. Phys. Chem. C, 2008, 112, 3334 CrossRef CAS; (b) L. D. Pachon, J. H. Maarseveen and G. Rothenberg, Adv. Synth. Catal., 2005, 347, 811 CrossRef; (c) A. Hartwig, M. Sebald and M. Kleemier, Polymer, 2005, 46, 2029 CrossRef CAS; (d) F. Miomandre, F. Chandezon, B. Lama, J. Besnardiere, M. Routier, A. Brosseau and P. Audebert, J. Nanopart. Res., 2011, 13, 879–886 CrossRef CAS; (e) M. Marini, F. Pilati and B. Pourabbas, Macromol. Chem. Phys., 2008, 209, 1374–1380 CrossRef CAS.
- J. Y. Kim, J. C. Park, H. Kang, H. Song and K. H. Park, Chem. Commun., 2010, 46, 439 RSC.
- (a) B. H. Lipshutz and B. R. Taft, Angew. Chem., Int. Ed., 2006, 45, 8235 CrossRef CAS; (b) H. Sharghi, R. Khalifeh and M. M. Doroodmanda, Adv. Synth. Catal., 2009, 351, 207 CrossRef CAS; (c) N. T. S. Phan and Ha. V. Le, J. Mol. Catal. A: Chem., 2011, 334, 130 CrossRef CAS.
- G. Molteni, C. L. Bianchi, G. Marinoni, N. Santod and A. Ponti, New J. Chem., 2006, 30, 1137 RSC.
- B. H. Lipshutz, D. M. Nihan, E. Vinogradova, B. R. Taft and Z. V. Boskovic, Org. Lett., 2008, 10, 4279 CrossRef CAS.
- I. S. Park, M. S. Kwon, Y. Kim, J. S. Lee and J. Park, Org. Lett., 2008, 10, 497 CrossRef CAS.
- E. A. Karakhanov, A. L. Maximov, Y. S. Kardasheva, V. A. Skorkin, S. V. Kardashev, V. V. Predeina, M. Y. Talanova, E. Lurie-Luke, J. A. Seeley and S. L. Cron, Appl. Catal., A, 2010, 385, 62 CrossRef CAS.
- J. L. Gole and M. G. White, J. Catal., 2001, 204, 249 CrossRef CAS.
- (a) D. J. Procter, J. Chem. Soc., Perkin Trans. 1, 2001, 335 RSC; (b) T. Kondo and T. A. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef CAS.
- D. D. Martino, M. C. Edler, G. La Regina, A. Cosuccia, M. C. Barbera, D. Barrow, R. I. Nicholson, G. Chiosis, A. Brancale, E. Hamel, M. Artico and R. Silvestri, J. Med. Chem., 2006, 49, 947 CrossRef.
- S. W. Kadlor, V. J. Kalish, J. F. Davies, B. V. Shetty, J. E. Fritz, K. Appelt, J. A. Burgess, K. M. Campanale, N. Y. Chirgadze, D. K. Clawson, B. A. Dressman, S. D. Hatch, D. A. Khalil, M. B. Kosa, P. P. Lubbehusen, M. A. Muesing, A. K. Patick, S. H. Reich, K. S. Su and J. H. Tatlock, J. Med. Chem., 1997, 40, 3979 CrossRef.
- (a) S. Balakumar, P. Thanasekaran, E. Rajkumar, K. John Adaikalasamy, S. Rajagopal, R. Ramaraj, T. Rajendran, B. Manimaran and K.-L. Lu, Org. Biomol. Chem., 2006, 4, 352 RSC; (b) E. Rajkumar and S. Rajagopal, Photochem. Photobiol. Sci., 2008, 7, 1407 RSC; (c) G. A. Gnanaraj, S. Rajagopal, C. Srinivasan and K. Pitchumani, Tetrahedron, 1993, 49, 4721 CrossRef CAS; (d) G. A. Gnanaraj, S. Rajagopal and C. Srinivasan, Tetrahedron, 1994, 50, 9447 CrossRef CAS.
- (a) L. Rout, T. K. Sen and T. Punniyamurthy, Angew. Chem., Int. Ed., 2007, 46, 5583 CrossRef CAS; (b) B. C. Ranu, A. Saha and R. Jana, Adv. Synth. Catal., 2007, 349, 2690 CrossRef CAS; (c) V. P. Reddy, A. Vijay Kumar, K. Swapna and K. Rama Rao, Org. Lett., 2009, 11, 1697 CrossRef CAS; (d) S. Bhadra, B. Sreedhar and B. C. Ranu, Adv. Synth. Catal., 2009, 351, 2369 CrossRef CAS.
- R. J. White, R. Luque, V. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481 RSC.
- G. Glaspell, H. M. A. Hassan, A. Elzatahry, V. Abdalsayed and M. S. El-Shall, Top. Catal., 2008, 47, 22 CrossRef CAS.
- P. Veerakumar, Z.-Z. Lu, M. Velayudham, K.-L. Lu and S. Rajagopal, J. Mol. Catal. A: Chem., 2010, 332, 128 CrossRef CAS.
- N. Perkas, Z. Zhong, J. Grinblat and A. Gedanken, Catal. Lett., 2008, 120, 19 CrossRef CAS.
- A. Abad, A. Corma and H. Garcia, Chem.–Eur. J., 2008, 14, 212 CrossRef CAS.
- J. M. Moreno, M. A. Aramendia, A. Marinas, J. M. Marinas and F. J. Urbano, Appl. Catal., B, 2007, 76, 34 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62 CrossRef.
- (a) S. Cheng, X. Wang and S. Y. Chen, Top. Catal., 2009, 52, 681 CrossRef CAS; (b) Y. Kobayashi and T. Sakuraba, Colloids Surf., A, 2008, 317, 756 CrossRef CAS.
- P. Pulkkinen, J. Shan, K. Leppanen, A. Kansakoski, A. Laiho, M. Jarn and H. Tenhu, ACS Appl. Mater. Interfaces, 2009, 1, 519 CAS.
- L. M. Kantam, S. V. Jaya, B. Sreedhar, M. M. Rao and B. M. Choudary, J. Mol. Catal. A: Chem., 2006, 256, 273 CrossRef.
- S. G. Alvarez and M. T. Alvarez, Synthesis, 1997, 413 CrossRef CAS.
- Y. Lee, J.-R. Choi, K. J. Lee, N. E. Stott and D. Kim, Nanotechnology, 2008, 19, 415604 CrossRef.
- (a) P. K. Khanna, S. Gaikwad, P. V. Adhyapak, N. Singh and R. Marimuthu, Mater. Lett., 2007, 61, 4711 CrossRef CAS; (b) X. Liu, S. Miao and B. Ji, J. Phys. Chem. Solids, 2007, 68, 1375 CrossRef CAS; (c) X. Zhang, H. Yin, X. Cheng, H. Hu, Q. Yu and A. Wang, Mater. Res. Bull., 2006, 41, 2041 CrossRef CAS.
- I. Haas, S. Shanmugam and A. Gedanken, J. Phys. Chem. B, 2006, 110, 16947 CrossRef CAS.
- C. Wu, B. P. Mosher and T. Zeng, J. Nanopart. Res., 2006, 8, 965 CrossRef CAS.
- (a) S. R. Wang, L. J. Zhu, Y. Y. Zhu, X. L. Ge and X. B. Li, Chin. Chem. Lett., 2011, 22, 362 CrossRef CAS; (b) Y.-Y. Zhu, S.-R. Wang, L.-J. Zhu, X.-L. Ge, X.-B. Li and Z.-Y. Luo, Catal. Lett., 2010, 135, 275 CrossRef CAS.
- H. H. Huang, F. Q. Yan, Y. M. Kek, C. H. Chew, G. Q. Xu, W. Ji, P. S. Oh and S. H. Tang, Langmuir, 1997, 13, 172 CrossRef CAS.
- (a) S. Chen, S. Hayakawa, Y. Shirosaki, E. Fujii, K. Kawabata, K. Tsuru and A. Osaka, J. Am. Ceram. Soc., 2009, 92, 2074 CrossRef CAS; (b) Z. Huang, F. Cui, H. Kang, J. Chen, X. Zhang and C. Xia, Chem. Mater., 2008, 20, 5090 CrossRef CAS; (c) S. Chen, A. Osaka, S. Hayakawa, K. Tsuru, E. Fujii and K. Kawabata, J. Sol-Gel Sci. Technol., 2008, 48, 322 CrossRef CAS.
- J. Liu, N. Ruffini, P. Pollet, V. Llopis-Mestre, C. Dilek, C. A. Eckert, C. L. Liotta and C. B. Roberts, Ind. Eng. Chem. Res., 2010, 49, 8174 CrossRef CAS.
- H. A. Orgueira, D. Fokas, Y. Isome, P. C.-M. Chan and C. M. Baldino, Tetrahedron Lett., 2005, 46, 2911 CrossRef CAS.
- D. Raut, K. Wankhede, V. Vaidya, S. Bhilare, N. Darwatkar, A. Deorukhkar, G. Trivedi and M. Salunkhe, Catal. Commun., 2009, 10, 1240 CrossRef CAS.
- F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Tetrahedron Lett., 2009, 50, 2358 CrossRef CAS.
- L. A. Belfiore and E. M. Indra, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 552 CrossRef CAS.
- (a) C. Aymonier, U. Schlotterbeck, L. Antonietti, P. Zacharias, R. Thomann, J. C. Tiller and S. Mecking, Chem. Commun., 2002, 3018 RSC; (b) K. Kim, B. H. Lee, J. W. Lee, H. K. Park and K. S. Shin, Langmuir, 2008, 24, 7178 CrossRef CAS; (c) S. Kidambi and M. L. Bruening, Chem. Mater., 2005, 17, 301 CrossRef CAS.
- A. M. Smith, H. Duan, M. N. Rhyner, G. Ruan and S. Nie, Phys. Chem. Chem. Phys., 2006, 8, 3895 RSC.
- (a) S. Jammi, S. Sakthivel, L. Rout, T. Mukherjee, S. Mandal, R. Mitra, P. Saha and T. Punniyamurthy, J. Org. Chem., 2009, 74, 1971 CrossRef CAS; (b) R. S. Schwab, D. Singh, E. E. Alberto, P. Piquini, O. E. D. Rodrigues and A. L. Braga, Catal. Sci. Technol., 2011, 1, 569 RSC.
- C.-K. Chen, Y.-W. Chen, C.-H. Lin, H.-P. Lin and C.-F. Lee, Chem. Commun., 2010, 46, 282 RSC.
- C. Gonzalez-Arellano, R. Luque and D. J. Macquarrie, Chem. Commun., 2009, 1410 RSC.
- (a) T. L. Pugh and W. Heller, J. Polym. Sci., 1960, 47, 219 CrossRef CAS; (b) A. B. R. Mayer and J. E. Mark, J. Macromol. Sci., Part A: Pure Appl. Chem., 1997, 34, 2151 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: SEM, XRD spectrum of the catalyst before and after usage, HRTEM images of catalyst A before and after usage, additional HRTEM images, AFM images, SEM images, BET analysis, UV-visible, 1H-NMR and 13C-NMR spectra of azides, 1,2,3-triazoles and C–S couple products. |
| This journal is © The Royal Society of Chemistry 2011 |