A reusable Mn(II)-dampy-MCM-41 system for single step amination of benzene to aniline using hydroxylamine
Sudarshan
Singha
and
K. M.
Parida
*
Colloids and Materials Chemistry Department, Institute of Minerals & Materials Technology, Bhubaneswar-751013, Orissa, India. E-mail: paridakulamani@yahoo.com; Fax: +91 674-2581637
First published on 7th September 2011
Abstract
Reaction methodology for C–H bond amination of benzene catalyzed by Mn(II)-dien (ampy) immobilized organo modified MCM-41 in the presence of hydroxylamine in acetic acid–water medium is described. The organic–inorganic hybrid heterogeneous catalyst achieves high catalytic activity and selectivity for one step amination of benzene. Characterization of the immobilized catalyst by powder X-ray diffraction (XRD), N2 adsorption–desorption, CP MAS NMR spectroscopy (13C and 29Si), Fourier transform infrared spectroscopy (FT-IR) and diffuse reflectance UV–Vis spectroscopy demonstrates the successful grafting of the complex into functionalized mesoporous silica and that the mesostructure has not been destroyed in the multistep synthetic procedure. The variation of reaction conditions such as solvent, temperature, time, catalyst concentration etc. is well demonstrated to achieve high catalytic activity. Moreover, the anchored catalyst can be recovered and reused for multiple cycles without appreciable loss in catalytic activity, which is an alternative to the conventional industrial process.
Introduction
The invention of an efficient and selective methodology for C–H bond oxidation possesses a formidable challenge in reaction design. A versatile protocol for both C–H bond amination and hydroxylation has become available in recent years. The vast majority of such processes are directed by attendant functional groups through covalent and noncovalent attachments.1,2 The power of these methods notwithstanding, high yielding and chemoselective intermolecular oxidation of C–H centers remains a most alluring problem of great potential reward.3 Methodologies have been recently developed for regioselective C–C,4 C–O5 or C–N6,7 bond formation that have found application in total synthesis.8Aniline is used as an important intermediate for preparation of dyes (such as aniline black, azo dyes, emeraldin etc.), N,N′-dialkyl-p-phenylene diamine (used to inhibit the oxidation of various organic materials which are normally subject to oxidative deterioration) and as a starting material for various organic syntheses.9 The existing method for production of aniline involves multistep reaction (nitration of an aromatic substrate followed by hydrogenation of the nitro group) which has many disadvantages (e.g. high energy requirement, time consuming, huge byproduct production causing a serious environmental hazard) and also disobeys the “greening” trends of global chemical manufacturing.10 Single step production of aniline via direct amination of benzene significantly improves the atomic efficiency, and is an attractive and challenging method from the point of view of both green chemistry and synthetic chemistry.
Hydrogen peroxide was found to be suitable for the direct amination of benzene on V–Ni/Al2O311 using ammonia as an aminating agent under mild conditions (low temperature, atmospheric pressure). However only modest yields were obtained. Therefore there is a great need for enhancement of both aniline yield and selectivity. Mantegazza et al.12 reported the direct synthesis of hydroxylamine with high yield by oxidation of ammonia with H2O2 in the presence of Ti-silicate at low temperature under atmospheric pressure. Upon suitable selection of solvent, no potential deep oxidation of ammonia occurs and no byproduct is formed. Thus, it provided a potential benign supply of hydroxylamine for the amination of benzene. Many attempts have been made to achieve liquid phase amination of benzene with hydroxylamine over a transition metal redox catalyst.13,14 Although the use of hydroxylamine in amination is not economically advantageous or environmentally benign at present, the study of the direct amination of benzene with hydroxyl amine will help in providing information for synthesizing aniline like molecules with ammonia and H2O2 in the future.
Transition metal functionalized MCM-41 has been reported to be an excellent catalyst for various organic synthesis and transformation reactions of industrial significance.15,16 Functionalization of MCM-41 by manganese leads to an effective heterogeneous catalyst for various oxidation reactions.17,18 In the recent years, the heterogenisation of transition metal complexes through immobilization onto solid supports especially MCM-41 has received great attention due to the inherent advantages of shape selectivity, easy separation and recycling of the catalyst, product purification and better handling properties.19 Complex encapsulation and methodology involving grafting and tethering procedures have shown several advantages over methods based on non-covalent interactions, such as electrostatic, π–π and hydrophobic/hydrophilic interactions and hydrogen bonding.20 In our previous work we have reported an Mn-MCM-41 mesoporous molecular sieve of benzene, where 68.5% conversion for benzene was observed.21 In continuation of our interest in exploring the higher conversion we employ a new protocol for immobilization of the Mn(II)-dien(ampy) complex onto chloropropyl modified MCM-41. The heterogeneous catalyst is robust enough to achieve a high catalytic performance for single step amination of benzene with NH2OH in water/acetic acid medium. Moreover, the anchored catalyst can be recycled multiple times without significant loss in catalytic activity.
Results and discussion
Powder X-ray diffraction
Fig. 1 shows the powder XRD patterns of MCM-41, CP-MCM-41, Mn(II)-dampy-MCM-41 and recycled catalyst (inset). The powder X-ray diffraction pattern of the parent MCM-41 shows a typical three-peak pattern with a very strong peak at 2θ ≈ 2.18° that corresponds to the d100 reflection and two other weaker peaks at 2θ ≈ 3.75° and 2θ ≈ 4.32° for d110 and d200 reflections respectively. All the peaks are well resolved indicative of good quality material. However, after organo-functionalization and metal complex loading there is a slight decrease in intensity with broadening of corresponding peaks indicating a slight disorder in the CP-MCM-41 and Mn(II)-dampy-MCM-41. The addition of an organic group probably distorts the regular liquid crystalline array of the template and lowers the long-range order of the MCM-41 mesostructure. The modest intensity reduction of reflections may be mainly due to contrast matching between the silicon frameworks of organic moieties which are located inside the framework of MCM-41.22 In the case of the recycled catalyst (Fig. 1(d) inset), all the peaks are retained in their respective position with respect to Mn(II)-dampy-MCM-41, suggesting that properties of the recycled catalyst remain unaltered.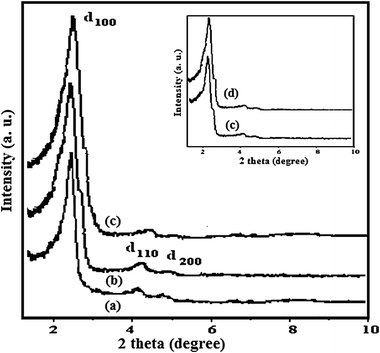 | ||
| Fig. 1 XRD spectra of(a) MCM-41, (b) CP-MCM-41, (c) Mn(II)-dampy-MCM-41 and (d) recycled catalyst (inset). | ||
N2 sorption studies
N2 adsorption–desorption isotherms of MCM-41, CP-MCM-41 and Mn(II)-dampy-MCM-41 are given in Fig. 2. Analysis of N2 adsorption and desorption data indicates a typical type-IV isotherm (defined by IUPAC) with a small hysteresis for neat MCM-41, CP-MCM-41 and Mn(II)-dampy-MCM-41, which is a characteristic for a mesoporous structure. The relative pressure at which pores are filling in the modified samples shifted to a lower value in comparison to the parent materials. Also a decrease in the BET specific surface area, pore volume and pore diameter was observed. The results are presented in Table 1. The nitrogen sorption study showed that the BET surface area of MCM-41 is 1280 m2 g−1 and the mesopore volume is 1.18 cm3 g−1. The average pore diameter is calculated to be 30.7 Å using the BJH method. All calculated values are in agreement with those reported for good quality mesoporous silica. The CP-MCM-41 shows less N2 uptake (BET surface area 989 m2 g−1) and pore volume 1.03 cm3 g−1 and Mn(II)-dampy-MCM-41 shows even lesser N2 uptake (BET surface area of 589 m2 g−1) and pore volume 0.68 cm3 g−1. From this, we can observe that lower pore volume and size distributions are obtained in the heterogeneous catalyst. It is due to the reason that the inner surface had been tailored with polar species and thus the adsorption of the nitrogen molecule is difficult. With the increase in bulkiness inside the pores, the hysteresis occurred at lower P/P0 indicating the decrease of pore size, which is a clear indication of immobilization of the Mn complex inside the pore channel of mesoporous silica.23 The loop at higher P/P0 (>0.9) is due to multilayer adsorption of the external surface area, but it does not lead to significant increase in surface area. | ||
| Fig. 2 N2 adsorption–desorption isotherms of MCM-41, CP-MCM-41 and Mn(II)-dampy-MCM-41. | ||
| Samples | BET surface area/m2 g−1 | Pore volume/cm3 g−1 | Pore diameter/Å |
|---|---|---|---|
| MCM-41 | 1280 | 1.18 | 30.7 |
| CP-MCM-41 | 989 | 1.03 | 26.1 |
| Mn(II)-dampy-MCM-41 | 589 | 0.68 | 20.6 |
13C CP-MAS NMR studies
The 13C CP-MAS NMR spectrum of CPTES functionalized MCM-41 is shown in Fig. 3(a). The sharp peak at 10.4 ppm is ascribed to the carbon atom bonded to silicon. The signal at 22.4 ppm corresponds to methylene carbon and the peak at 50 ppm can be attributed to the carbon atom attached to the chlorine atom. The 13C CP MAS NMR spectra of complex immobilized MCM-41 (Fig. 3(b)) samples show peaks corresponding to the carbon atom of chloropropyl functionalization and both the aliphatic and aromatic carbons of the metal complex. | ||
| Fig. 3 13C CP-MAS NMR spectra of (a) CP-MCM-41, (b) Mn(II)-dampy-MCM-41. 29Si CP-MAS NMR spectra of (c) MCM-41 and (d) CP-MCM-41. | ||
29Si CP-MAS NMR studies
The degree of functionalization i.e. the covalent linkage between the silanol groups and the organic moiety on the mesostructured materials also can be monitored by means of 29Si CP-MAS NMR spectroscopy. The spectrum of silicous MCM-41 (Fig. 3(c)) generally exhibits three resonance peaks at δ −110, −101, and −92 ppm corresponding to Q4 [siloxane, (SiO)4Si], Q3 [single silanol, (SiO)3Si(OH)] and Q2 [geminal silanol, (SiO)2Si(OH)2] sites of the silica framework respectively. Covalent binding makes the Q2 signal disappear, decreases Q3 and concomitantly increases the Q4 intensity, which is due to consumption of isolated Si–OH and geminal silanediols during the condensation process. Two resonance peaks due to the Si environments of Q4 (δ = −110 ppm) and Q3 (δ = −102 ppm) can be seen in the organic group modified silica. In addition to these two peaks, the sample displays two more resonance peaks at δ = −68 ppm, assigned to T3 [C–Si(OSi)3], and at −57 ppm, attributed to T2 [C–Si(OSi)2(OH)] respectively. The existence of T3 confirms that MCM-41 has been modified by organic moieties.24,25 The appearance of a Q3 signal indicates the presence of some residual noncondensed OH groups attached to the silicon atom. The 29Si CP-MAS NMR provides direct evidence that the hybrid CP-MCM-41 sample consists of a highly condensed siloxane network with an organic group covalently bonded to the mesoporous silica (Fig. 3(d)). These two peaks due to the Si atoms of different environments in the organosilane CP-MCM-41 also revealed that both the synthesis process and surfactant-extraction treatment did not cause cleavage of the Si–C bonds.FTIR studies
FTIR has been shown to be a powerful characterization technique for monitoring multi-step-assembly of the complex into the mesoporous silica. The FTIR spectra of the MCM-41 (Fig. 4(a)) showing specific bands at around 1110, 803 and 467 cm−1 assigned to characteristic vibration of the mesoporous framework (Si–O–Si) and a broad peak around 3450 cm−1 may be attributed to surface silanol and adsorbed water molecules, whose deformational vibration mode is assigned to the band at 1623–1640 cm−1. It can be analyzed that the band appearing near 2900 cm−1 is due to the C–H stretching of the functionalized chloropropyl group (which does not appear in the parent MCM-41 spectra). After anchoring the ligand on to the functionalized MCM-41 surface, a band near 3360 cm−1 appears which results due to the N–H stretching of the secondary amine. The C–N stretching in the range 1250–1020 cm−1 is masked by the Si–O–Si characteristic bands. After metalation all the bands remain intact (as present in the ligand), only the N–H band shifts towards lower frequency indicating the formation of a complex. In case of recycled catalysts (Fig. 4(e)) none of the vibrations change significantly compared to Mn(II)-dampy-MCM-41 (Fig. 4(d)). This suggests existence of all the properties in recycled catalyst compared to fresh heterogeneous catalysts. | ||
| Fig. 4 FT-IR spectra of (a) MCM-41, (b) CP-MCM-41, (c) ligand modified-MCM-41 (d) Mn(II)-dampy-MCM-41 and (e) recycled catalysts. | ||
UV-vis spectroscopy
The diffuse-reflectance UV-vis spectra of (a) CP-MCM-41, (b) dien (ampy) modified CP-MCM-41 and (c) Mn(II)-dampy-MCM-41 are shown in Fig. 5. Chloropropyl modified mesoporous silica does not show any significant characteristic absorption band. The spectrum for ligand modified silica showing bands at 286 and 360 nm could be attributed to intra-ligand (π–π* and n–π*) charge transfer transition. After complexation the intensities of the band diminished due to the coordination of the metal to the site of the ligand. Mn(II)-dampy-MCM-41 shows additional peaks in the region of 550–600 nm which may be assigned to d–d transition of the metal. The electronic spectra confirmed the immobilization of the Mn(II)-dien(ampy) complex on the organo modified silica support.
Scanning electron microscopy
Fig. 6 shows the SEM images of (a) MCM-41, (b) CP-MCM-41 and (c) Mn(II)-dampy-MCM-41. The SEM image of MCM-41 shows spherically uniform morphology. In the case of chloro functionalized MCM-41 the spherical shape slightly tends to elongate. However the complex immobilized sample reveals slight agglomerisation of particles but morphology does not change appreciably compared to former samples. | ||
| Fig. 6 SEM images of (a) MCM-41 (b) CP-MCM-41 and (c) Mn(II)-dampy-MCM-41. | ||
Reaction pathway
A free-radical mechanism has been proposed for amination reaction using N-chloroalkyl amines, hydroxylamine-o-sulfonic acid and alkyl hydroxylamine as the aminating agents catalyzed by redox metal ions such as Ti3+, Fe2+ and Cu+.26 In our previous work, a similar type of free radical mechanism has been reported for amination of benzene catalyzed by Mn-MCM-41. Kuznetsova et al.27 proposed a free-radical mechanism, considering the protonated amino radical (˙NH3+) as the active aminating species, for amination of benzene and toluene. An immobilized heterogeneous catalyst along with acidic media promotes the generation of a protonated amino (˙NH3+) radical by reduction of hydroxylamine. In the subsequent step the protonated amino radical reacts with benzene to give protonated aminocyclohexadienyl intermediates. Oxidation of unstable intermediates leads to production of aniline along with regeneration of the catalyst (Scheme 1). Along with the ˙NH3+ radical, ˙NH2 is also formed as per the predicted mechanism. However the solvent effect indicates the favored participation of the ˙NH3+ in the catalytic reaction. Hydroxylamine exists as HONH3+ in weak acid solution and normally acts as a reducing agent.28 The protonated hydroxylamine is prone to undergo a disproportionation as per eqn (5) in Scheme 1. In addition, the chemical structure of NH2OH is suitable to interact with the manganese complex replacing the labile groups and thus making NH2OH unstable. Thus the present catalytic amination carried out in HOAc–H2O medium is likely to be accompanied by the decomposition of hydroxylamine, by either disproportionation and/or its interaction with the manganese species. In both the reactions, gaseous NO2 would be produced. So decomposition of hydroxylamine can be calculated by measuring the amount of gaseous NO2 produced over time during the reaction period. With benzene, after suitable work-up, aniline was the only identified product.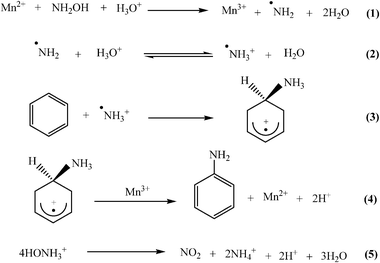 | ||
| Scheme 1 The possible reaction pathway. | ||
The influence of various reaction parameters such as different reaction times, temperatures, solvents, amount of hydroxylamine and catalyst loading has been investigated to optimize the reaction conditions.
Effect of solvent
Suitable selection of solvent is quite essential as it plays an immense role during the amination reaction. Solvents such as acetone, acetonitrile, acetic acid, water and acetic acid–water medium were tried for the reaction. In acetone, acetonitrile, water and acetic acid medium only 4.6%, 10.2%, 20.3% and 53.4% benzene conversion took place. The highest conversion of 74.6% was obtained using acetic acid–water (70 vol% acetic acid) as the solvent, which indicates that there is some influence of acidic medium on generation of the ˙NH3+ radical. The pKa value of ˙NH3+ is 3.7526 indicating the ease of formation of the ˙NH3+ radical rather than the neutral amino radical (˙NH2) in the reaction medium. The increase in the conversion of benzene was observed with the increase in acetic acid concentration up to 70 vol% but a reverse effect was observed with further increase in the concentration. This may be attributed to the fact that with this range of concentration acetic acid exhibits good solubility and favorable acidity for highest decomposition of hydroxylamine. This results in highest conversion of benzene and 100% selectivity for aniline.Effect of reaction temperature
The reaction was carried out between 30 °C and 80 °C keeping other parameters fixed. The experimental results for benzene conversion and aniline selectivity are shown in Fig. 7. No conversion was observed up to 30 °C and no decomposition of hydroxylamine was observed, indicating that the amination process is associated with decomposition of hydroxylamine. There is a sudden increase in the conversion from 21.1% to 74.6% with increase in temperature from 50 °C to 70 °C. The sharp decrease in conversion of benzene to aniline at 80 °C may presumably be due to the vaporization of benzene.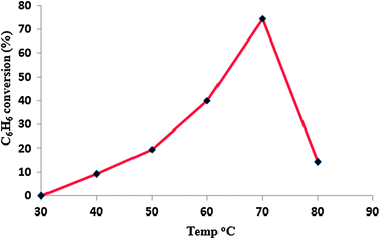 | ||
| Fig. 7 Effect of temperature on amination of benzene. Benzene = 11.25 mmol, NH2OH = 11.25 mmol, acetic acid = 7.5 ml (70 vol%), catalyst amount = 0.05 g, time=2 h. | ||
Effect of hydroxylamine amount
Fig. 8 shows the variation of % conversion of benzene with the amount of hydroxylamine. On variation of the nNH2OH/nbenzene molar ratio from 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1 the conversion of benzene increases from 65% to 71.2%, aniline selectivity from 95% to 100% and hydroxylamine from 65.8% to 88.6%. Again with a further increase in the molar ratio to 2:1 the benzene conversion increases up to 81% but the selectivity for aniline and hydroxylamine decreases considerably which can be analyzed from the figure.
:1 to 1:1 the conversion of benzene increases from 65% to 71.2%, aniline selectivity from 95% to 100% and hydroxylamine from 65.8% to 88.6%. Again with a further increase in the molar ratio to 2:1 the benzene conversion increases up to 81% but the selectivity for aniline and hydroxylamine decreases considerably which can be analyzed from the figure.
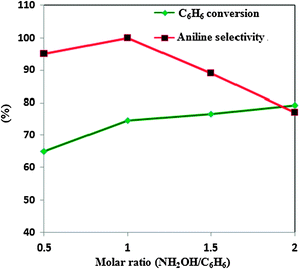 | ||
| Fig. 8 Effect of the molar ratio of hydroxylamine/benzene on amination of benzene. Benzene = 11.25 mmol, acetic acid = 7.5 ml (70 vol%), catalyst amount = 0.05 g, temp. = 70 °C, time = 2 h. | ||
Effect of immobilization
Amination of benzene was also carried out using the “neat” Mn(II)-dien(ampy) complex containing the same amount of manganese as in the case of the immobilized complex (Table 2). It is evident from the table that the conversion increases on immobilization of the complex. Mn(II)-dien(ampy) in the solution may presumably form dimers that inhibit the catalytic activity. But the Mn(II)-dien(ampy) molecules are isolated and well separated from each other since they are anchored to the wall of MCM-41, which is responsible for the increase in conversion.Catalytic reusability test
Reutilization is one of the greatest advantages of a heterogeneous catalyst, which can also provide useful information about the immobilization process and catalytic stability along the catalytic cycles. To address the issue, a series of tests were undertaken. To test if the metal is leached out from the solid catalyst during reaction, the liquid phase of the reaction mixture is collected by filtration at the reaction temperature (70 °C). Atomic absorption spectrometric analysis of the liquid phase of the reaction mixture thus collected by filtration confirms the absence of metal ions in the liquid phase. These experiments clearly demonstrate that manganese does not leach out from the solid catalyst during amination reaction. After the catalytic reactions were completed, a solid catalyst was recovered by filtration and washed with ethanol several times and dried in open air. The recovered catalyst was then subjected to XRD, FT-IR spectroscopy. Comparison of XRD patterns and IR spectra of fresh and recovered catalysts convincingly demonstrates that the structural integrity of the complex intercalated MCM-41 remains unaltered after the amination reaction. Notably, AAS analysis (Table 3) confirmed that the manganese loading of the recycled catalyst was not obviously reduced after four cycles; also the catalytic activity was not decreased compared to that of the fresh one.| Run | Conversion (%) of BZ | Selectivity (%) of AN | Wt% of Mn from AAS |
|---|---|---|---|
| a Reaction conditions: temp =70 °C, benzene = 11.25 mmol, catalyst amount = 0.05 g, acetic acid = 7.5 ml (70 vol%), time = 2 h, NH2OH = 11.25 mmol, AN = aniline, BZ = benzene. | |||
| Fresh | 74.6 | 100 | 0.42 |
| 1 | 74.1 | 100 | 0.42 |
| 2 | 73.3 | 100 | 0.41 |
| 3 | 71.9 | 100 | 0.41 |
| 4 | 70.2 | 100 | 0.41 |
Experimental section
Materials and reagents
Tetraethylorthosilicate (TEOS), cetyltrimethylammoniumbromide (CTAB), 3-chloropropyl triethoxysilane (CPTES), pyridine 2-carbaldehyde, diethylenetriamine, and Mn(OAc)2 were obtained from Aldrich. Sodium hydroxide, sodium borohydride (NaBH4), benzene, and hydroxylamine hydrochloride (NH2OH·HCl) were obtained from Merck. All the solvents were of AR grade, and were procured from SD fine Chemicals, India, and were distilled and dried before use. Ethanol was dried using a molecular sieve 5A prior to its use.Catalyst preparation
:8.16:1.05:2.55:4.857) based on the molar ratio. The mixture of CTAB (2.0 g, 5.49 mmol), 2.0 M of NaOH (aq) (7.0 ml, 14.0 mmol) and H2O (480 g, 26.67 mol) was heated at 80 °C for 30 min to reach pH 12.3. To this clear solution TEOS (9.34 g, 44.8 mmol) and CPTES (1.31 g, 5.75 mmol) were added sequentially and rapidly via injection. Following the injection, a white precipitate was observed after 3 min of stirring. The reaction temperature was maintained at 80 °C for 2 h. The products were isolated by hot filtration, washed with copious amounts of water and ethanol and dried under vacuum.
With this dried product, an acid extraction process was performed by adding 1.0 g of the material in a mixture of 100 ml ethanol and 1.0 ml concentrated hydrochloric acid at 60 °C for 6 h. The resulting surfactant removed product was filtered and washed with water and ethanol and dried under vacuum. The final product was denoted as Cl-MCM-41.
:30) in the liquid phase. The solid was separated by filtration. By evaporation of the solvent, a pale yellow solid was obtained. Yield of the product obtained was found to be 79%. The ligand can be abbreviated as dien (ampy).
Immobilization of the manganese complex
The experiment was carried out in a nitrogen atmosphere in order to avoid a real oxidation. To a suspension of freshly dried Cl-MCM-41 (1 g) in dry toluene (40 ml), the ligand (0.1 g) in dry toluene (10 ml) was added and the resulting solution was refluxed for 3 h at 50 °C. The faint yellow colored solid was separated, washed several times with ethanol to get rid of the unreacted ligands on the external surface of Cl-MCM-41 and dried in vacuum for 24 h. The ligand gets attached to the MCM-41 through the spacer by the nucleophilic displacement of chloride of Cl-MCM-41 by the basic amino group of the ligand. Then to a suspension of this solid (1 g) in dry ethanol (40 ml), a solution of manganese acetate (0.1 g) in dry ethanol (25 ml) was added and refluxed for 3 h. The solution immediately turned light brown indicating the formation of the manganese complex. The light brown solid was separated by filtration, was dried and soxhlet extracted with dry ethanol and acetonitrile to remove excess unreacted manganese from the surface. The manganese content in Si-MCM-41 was estimated to be 0.42 wt% by AAS. The resulting product is denoted as Mn(II)-dampy-MCM-41. The schematic representation of preparation is depicted in Scheme 2. 13C CP MAS NMR (100.62 MHz, CDCl3, ppm): δ = 9, 22, 27, 43, 50, 53, 60, 110, 127, 161, 165. The homogeneous Mn(II)-dampy complex was prepared by refluxing equimolar amounts of manganese acetate with the prepared dien (ampy) ligand. Fig. 9 shows the stick representation of the homogeneous Mn(II)-dampy complex. | ||
| Scheme 2 Mechanism of immobilization of the complex in MCM-41. | ||
 | ||
| Fig. 9 Stick model representation of the neat manganese complex. | ||
Physico-chemical characterizations
Powder X-ray diffraction (XRD) patterns of the samples were obtained on a Rigaku D/Max III VC diffractometer with Cu Kα radiation at 40 kV and 40 mA in the range of 2θ = 0°–10°. The scanning rate was 2° min−1. Nitrogen adsorption–desorption isotherms were measured at liquid nitrogen temperature using ASAP 2020 (Micromeritics). Samples were outgassed at 100 °C for 3 h to evacuate the physically adsorbed moisture before measurement. Specific surface area was calculated using the BET method. Solid-state 13C and 29Si cross-polarization magic-angle spinning NMR spectra were recorded at 100.58 and 79.46 MHz, respectively, using a Bruker Avance 400 MHz spectrometer.The FTIR spectra of the samples were recorded using Varian 800-FTIR in a KBr matrix in the range of 4000–400 cm−1. The co-ordination environments of the samples were examined by diffuse reflectance UV–Vis spectroscopy. The spectra were recorded in a Varian-100 spectrophotometer in the wavelength range of 200–800 nm in the BaSO4 phase. Scanning electron microscopy (SEM) images were obtained using a HITACHI 3400N microscope. The samples were placed on a copper tape and then coated with a thin layer of platinum (layer thickness 3 nm) using a sputter coater. The Mn loading and leaching of the reaction solution were determined by atomic absorption spectroscopy (AAS) with a Perkin-Elmer Analyser 300 using an acetylene (C2H2) flame. Chem 3D software was adopted to elucidate the ball–stick representation of the homogeneous Mn(II)-dampy complex.
Catalytic amination reaction
The catalytic amination reaction was carried out in a thermostatic two-necked round bottomed flask fitted with a reflux condenser at atmospheric pressure. In a typical experiment, 0.05 g of the catalyst and 11.25 mmol of NH2OH were loaded into a reaction flask containing 7.5 ml of 70 vol% acetic acid and the mixture was stirred for about 30 min at 30 °C. Then 1 ml of benzene (11.25 mmol) was introduced, and the reaction was performed at 70 °C. The resulting solution was cooled to room temperature and neutralized by saturated solution of sodium bicarbonate. The organic compound was extracted with diethyl ether and analyzed by gas chromatography, Shimadzu, GC-2010, equipped with a capillary column (ZB-1, 30 m length, 0.53 mm ID and 3.0 μm film thickness), using a flame ionization detector (FID). m-Toluidine was used as an internal standard to quantify the aniline produced. The schematic representation of the reaction is given in Scheme 3. | ||
| Scheme 3 The catalytic amination reaction. | ||
Decomposition of hydroxylamine was carried out at atmospheric pressure in argon flow with a flow rate of 25 ml min−1. The gas product that formed during the decomposition was analyzed by GC (3 m × 2.3 mm porapak Q column, TCD detector) after being dried by CaO.
The conversion and selectivity were calculated as follows:
Conversion (mol%) = {(initial mol% − final mol%)/initial mol%} × 100
Aniline selectivity = {(GC peak area of aniline)/(GC peak area of all products)} × 100
Hydroxylamine selectivity = {(GC peak area of hydroxylamine)/(GC peak area of all products)} × 100
Conclusions
We have found that the heterogenized manganese complex shows significant increase in amination activity under less acidic conditions. The employment of a chelating ligand has provided the major driving force for the evaluation of an active metal catalyst, which in turn promotes the catalytic activity. In this piece of work we got nearly 6% more conversion compared to our previous work (Mn-MCM-41), which is a better alternative from the synthetic view point. This would lead to design more number of metal complex immobilized heterogeneous catalysts for the single step production of aniline. The highly predisposed ligands allow the manganese centers to accommodate at least two exogenous ligands in the cis fashion, and they create thermodynamically and kinetically stable complexes, which are more resistant to metal decomplexation. The stability of the catalyst has also been demonstrated convincingly by conducting four successive runs without appreciable loss of reactivity.Acknowledgements
The authors are extremely thankful to Professor B.K. Mishra, Director, IMMT, Bhubaneswar 751013, Orissa, India, for his constant encouragement and permission to publish the paper. The authors are also thankful to Department of Science and Technology, New Delhi, for financial support.Notes and references
- C. G. Espino, J. Du Bois and P. A. Evans, Modern Rhodium-Catalyzed Organic Reactions, Wiley-VCH, Weinheim, 2005, 379 Search PubMed.
- K. J. Fraunhoffer, N. Prabagaran, L. E. Sirois and M. C. White, J. Am. Chem. Soc., 2006, 128, 9032 CrossRef CAS.
- M. M. Diaz-Requejo, T. R. Belderrain, M. C. Nicasio, S. Trofimenko and P. J. Perez, J. Am. Chem. Soc., 2003, 125, 12078 CrossRef CAS.
- F. Kakiuchi, S. Kan, K. Igi, N. Chatani and S. Murai, J. Am. Chem. Soc., 2003, 125, 1698 CrossRef CAS.
- L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542 CrossRef CAS.
- J. L. Brice, J. E. Harang, V. I. Timokhin, N. R. Anastasi and S. S. Stahl, J. Am. Chem. Soc., 2005, 127, 2868 CrossRef CAS.
- C. G. Espino, P. M. Wehn, J. Chow and J. Du Bois, J. Am. Chem. Soc., 2001, 123, 6935 CrossRef CAS.
- J. A. Johnson, N. Li and D. Sames, J. Am. Chem. Soc., 2002, 124, 6900 CrossRef CAS.
- L. Schmerling, US Patent, 2,948,755, 1960 Search PubMed.
- R. T. Baker and W. Tumas, Science, 1999, 284, 1477 CrossRef CAS.
- Y. S. Xia, L. F. Zhu, G. Y. Li and C. W. Hu, Acta Phys–Chim. Sin., 2005, 21, 1337 CAS.
- M. A. Mantegazza, G. Leofanti, G. Petrini, M. Padovan, A. Zeccina, S. Bordiga and V. C. Corberan, in New Developments in Selective Oxidation,Elsevier, ed. S. V. Bellon, New York, 1994, 51 Search PubMed.
- N. I. Kuznetsova, L. I. Kuznetsova, L. G. Detusheva, V. A. Likholobov, G. P. Pez and H. Cheng, J. Mol. Catal. A: Chem., 2000, 161, 1 CrossRef CAS.
- L. F. Zhu, B. Guo, D. Y. Tang, X. K. Hu, G. Y. Li and C. W. Hu, J. Catal., 2007, 245, 446 CrossRef CAS.
- S. Velu, L. Wang, M. Okazaki, K. Suzuki and T. Tomura, Microporous Mesoporous Mater., 2002, 54, 113 CrossRef CAS.
- I. Sobczak, M. Ziolek, M. Renn, P. Decyk, I. Nowak, M. Daturi and J. C. Lavalley, Microporous Mesoporous Mater., 2004, 74, 23 CrossRef CAS.
- Q. Zhang, Y. Wang, S. Itsuki, T. Shishido and K. Takehira, J. Mol. Catal. A: Chem., 2002, 188, 189 CrossRef CAS.
- S. Vetrivel and A. Pandurangan, J. Mol. Catal. A: Chem., 2006, 246, 223 CrossRef CAS.
- S. Ray, S. F. Mapolie and J. Darkwa, J. Mol. Catal. A: Chem., 2007, 267, 143 CrossRef CAS.
- Q. H. Fan, Y. M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385 CrossRef CAS.
- K. M. Parida, S. S. Dash and S. Singha, Appl. Catal., A, 2008, 351, 59 CrossRef CAS.
- M. Kruk, M. Jaroniec, Y. Sakamoto, O. Terasaki, R. Ryoo and C. H. Ko, J. Phys. Chem. B, 2000, 104, 292 CrossRef CAS.
- H. Yang, G. Zhang, X. Hong and Y. Zhan, J. Mol. Catal. A: Chem., 2004, 210, 143 CrossRef CAS.
- D. Jiang, Q. Yang, H. Wang, G. Zhu, J. Yang and C. Li, J. Catal., 2006, 239, 65 CrossRef CAS.
- D. Jiang, Q. Yang, J. Yang, L. Zhang, G. Zhu and C. Li, Chem. Mater., 2005, 17, 6154 CrossRef CAS.
- A. Citterio, A. Gentile, F. Minisci, V. Navaovini, M. Sevravalle and S. Ventura, J. Org. Chem., 1984, 49, 4479 CrossRef CAS.
- N. I. Kuznetsova, L. I. Kuznetsova, L. G. Detusheva, V. A. Likholobov, G. P. Pez and H. Cheng, J. Mol. Catal. A: Chem., 2000, 161, 1 CrossRef CAS.
- C. Wei, S. R. Saraf, W. J. Roger and M. S. Mannan, Thermochim. Acta, 2004, 421, 1 CrossRef CAS.
- D. Brunel, Microporous Mesoporous Mater., 1999, 27, 329 CrossRef CAS.
- P. Adao, J. C. Pessoa, R. T. Henriques, M. L. Kuznetsov, F. Avecilla, M. R. Maurya, U. Kumar and I. Correia, Inorg. Chem., 2009, 48, 3542 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |