In situ green synthesis of Au nanostructures on graphene oxide and their application for catalytic reduction of 4-nitrophenol†
Yingwei
Zhang
a,
Sen
Liu
a,
Wenbo
Lu
a,
Lei
Wang
a,
Jingqi
Tian
ab and
Xuping
Sun
*a
aState Key Lab of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022 Jilin, China. E-mail: sunxp@ciac.jl.cn; Fax: +86 431-85262065; Tel: +86 431-85262065
bGraduate School of the Chinese Academy of Sciences, Beijing 100039, China
First published on 18th July 2011
Abstract
In this communication, we develop a relatively green, and environment friendly route for the synthesis of Au nanostructures on tannic acid (TA)-functionalized graphene oxide (GO) using TA as a reducing and immobilizing agent. The morphologies of Au nanostructures can be controlled by the amount of HAuCl4 used. The resultant Au nanostructures/GO nanocomposites exhibit good catalytic activity toward 4-nitrophenol (4-NP) reduction and the GO supports also enhance the catalytic activityvia a synergistic effect.
During the past years, considerable attention from both fundamental and applied research has been paid to synthesizing and characterizing metal nanoparticles, particularly Au and Ag, due to their unique physical and chemical properties compared to bulk metals and their very important applications in catalysis.1 Various methods for the preparation of such nanoparticles have been developed over the past years;2 however, the chemical reduction of metal ions in a solution is still the most common preparative route. Unfortunately, unprotected metal colloids are susceptible to irreversible aggregation in solution due to their small size, resulting in a remarkable reduction in their catalytic activities. To address these disadvantages, one of the effective strategies is the introduction of metal nanoparticles on/into less expensive solid supports (such as polymers, carbon, metal oxides and so on) to form composite catalysts.3 Up to now, there have been many efforts toward assembling Au nanoparticles on/into many kinds of supports with different nanostructures.4 Among these nanostructures, two-dimensional (2D) plate-like structured graphene and its derivative, graphene oxide (GO), with high surface areas have been attractive choices as promising candidates for supports.5 Currently, graphene has been successfully utilized as a support to disperse and stabilize metal nanoparticles, while few reports of water-soluble GO used as supports are available.6 Compared to graphene, GO should be a better support for the fabrication of water-soluble nanoparticles/2D nanocarbons due to its good dispersion in aqueous solution for applications.
Tannic acid (TA; Fig. S1, ESI†) as a water-soluble, phenolic hydroxyl-rich compound is widely present in woods such as oak, walnut and mahogany and has been used as a common mordant in industry. Recently, the π–π interactions between the aromatic rings of TA and GO sheets have been used to disperse GO sheets in aqueous medium successfully.7 In this communication, we report on our recent finding that Au nanostructures can be in situ reduced and immobilized on TA-functionalized GO through a green, cost-effective strategy with the use of TA as a reducing and immobilizing agent. The morphologies of Au nanostructures thus formed can be controlled by the amount of gold salts used. It suggests that the resultant Au nanostructures/GO nanocomposites exhibit good activities in catalyzing the reduction of 4-nitrophenol (4-NP) by NaBH4 and the GO support has a synergistic effect to enhance the catalytic activity of Au catalysts.
The successful preparation of Au nanoparticles/GO nanocomposites (see ESI† for preparation details) was confirmed by UV-vis absorption spectra, as shown in Fig. 1. The UV-vis spectrum of TA shows two peaks at 214 and 274 nm (curve a), which are assigned to π–π* and n–π* transition, respectively.7 The UV-vis spectrum of GO shows two peaks at 230 nm and 300 nm (curve b). The TA/GO shows the superimposed absorbance bands of TA and GO at 220 nm (curve c). After the addition of 30 μL HAuCl4 into the suspension of TA/GO, a new absorbance band appears at 522 nm characteristic of the colloidal gold surface plasmon resonance band, indicating the formation of gold nanoparticles (curve d). Bayberry tannin as a phenolic hydroxyl-rich compound has recently been successfully used as a reducing and stabilizing agent for the synthesis of Au nanoparticles and Pd nanoparticles.8 The spontaneous formation of Au nanoparticles in our present study can also be attributed to the direct redox between TA and Au3+, where Au3+ is reduced to metallic Au by the phenolic hydroxyls of TA, which is evidenced by the blue shift of the superimposed absorbance bands of TA/GO from 220 nm to 223 nm due to the decreased absorbance of TA.
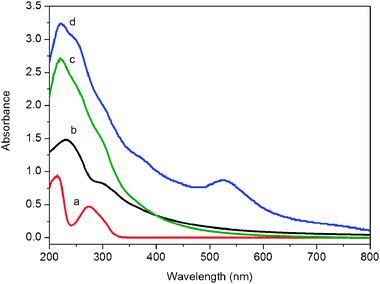 | ||
| Fig. 1 UV-vis absorption spectra of aqueous dispersions of TA (a), GO (b), TA/GO (c), and Au nanoparticles/GO (d). | ||
Fig. 2 shows the TEM images of the products thus formed (see ESI† for preparation details). It is seen that well-dispersed TA-functionalized GO sheets are observed in the suspension of TA/GO, as shown in Fig. 2a. After the addition of 30 μL of HAuCl4 into the suspension of TA/GO, it is seen that the GO sheet has been decorated with many small Au nanoparticles about several nanometres to 20 nm in diameter (Fig. 2b). When 60 μL of HAuCl4 was added into the suspension of TA/GO, Au nanoparticles with increased sizes of ca. 100 nm and a small amount of Au nanoplates are observed on the surface of GO (Fig. 2c). When the volume of HAuCl4 was further increased up to 100 μL, more triangular and hexagonal Au nanoplates with increased edge sizes are obtained on the surface of GO and some small Au nanoparticles are also observed, as shown in Fig. 2d. Those above results indicate that with the increase in the amount of HAuCl4, Au nanostructures with controllable morphologies from nanoparticles to nanoplates can be prepared on GO sheets with the use of TA as the reducing and immobilizing agent. Scheme 1 illustrates the noncovalent functionalization of GO by TA and the subsequent in situ synthesis of Au nanostructures with controllable morphologies on GO.
 | ||
| Fig. 2 TEM images of TA/GO (a), Au nanostructures/GO obtained with the use of 30 μL (b), 60 μL (c), and 100 μL (d) of HAuCl4. | ||
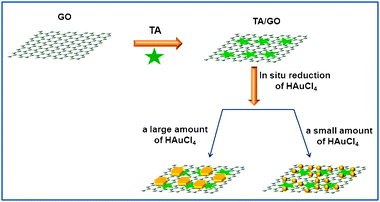 | ||
| Scheme 1 A scheme (not to scale) to illustrate the noncovalent functionalization of GO by TA and the subsequent preparation of Au nanostructures/GO nanocomposites by in situ chemical reduction of gold salts. | ||
It is well known that 4-aminophenol (4-AP) is very useful and important in many applications that include analgesic and antipyretic drugs, photographic developer, corrosion inhibitor, anticorrosion lubricant, and so on.9 The reduction of 4-NP by NaBH4 in the presence of noble metal nanoparticles as catalysts has been intensively investigated for the efficient production of 4-AP.10 Therefore, the reduction of 4-NP to 4-AP with an excess amount of NaBH4 was used as a model system to quantitatively evaluate the catalytic activities of the resultant Au nanostructures/GO nanocomposites. In the absence of our catalysts, the mixtures of 4-NP and NaBH4 show a strong adsorption peak at 400 nm, corresponding to the 4-NP ions under alkaline conditions.11 After the catalysts of Au nanostructures/GO nanocomposites were added into the reaction system, the reduction process was monitored by measuring the time-dependent adsorption spectra of the reaction mixture solution. As shown in Fig. 3, the absorption intensity of 4-NP at 400 nm decreases quickly with time, accompanied by the appearance of the new peaks of 4-AP at 230 nm and 300 nm, indicating conversion of 4-NP to 4-AP.12 It should be noted that the reduction of 4-NP by NaBH4 was finished within 18 minutes upon the addition of Au nanoparticles/GO (Fig. 3a), with the observation of a fading and ultimate bleaching of the yellow-green color of the reaction mixture in aqueous solution. However, a longer reaction time of 75 minutes was required to achieve the full reduction of 4-NP by NaBH4 using Au nanoplates/GO as catalysts (Fig. 3b). We also investigated the effects of the GO support on the catalytic activity. A reaction time of 27 minutes was required to achieve the full reduction of 4-NP by NaBH4 using Au nanoparticles alone (TA-reduced AuNPs, see ESI† for preparation details) as catalysts (Fig. 3c and Fig. S2, ESI†). Due to the presence of large excess of NaBH4 compared to 4-NP, the rate of reduction is independent of the concentration of NaBH4, and the reaction could be considered pseudo-first-order with respect to the concentration of 4-NP.13 Hence, ln(Ct/C0) versus time can be obtained based on the absorbance as the function of time, and good linear correlations are observed, as shown in Fig. 3d, suggesting that the reactions follow pseudo-first-order kinetics. Then, the kinetic reaction rate constants (defined as kapp) are estimated from the slopes of the linear relationship to be 18.8 × 10−2 min−1 (Au nanoparticles/GO), 12.2 × 10−2 min−1 (Au nanoparticles) and 4.67 × 10−2 min−1 (Au nanoplates/GO), respectively. These results clearly indicate the Au nanoparticles/GO composites have higher catalytic activity for the reduction of 4-NP than the Au nanoplates/GO composites, due to the smaller sizes and larger active surface areas of Au nanoparticles compared with Au nanoplates. It is also important to point out that the Au nanoparticles/GO composites show relatively higher catalytic activity than Au nanoparticles alone. Such observation indicates that the GO support may play an active part in the catalysis, yielding a synergistic effect. The synergistically enhanced catalytic activity may be explained as follows: given that 4-NP is π-rich in nature, it is expected that 4-NP can be adsorbed onto GO via π–π stacking interactions. Such adsorption provides a high concentration of 4-NP near to the Au nanoparticles on GO, leading to highly efficient contact between them. In contrast, without a highly adsorbent GO support, 4-NP must collide with Au nanoparticles by chance, and remains in contact for the catalysis to proceed. When this is not achieved, 4-NP will pass back into solution and can only react further when it collides with Au nanoparticles again.14
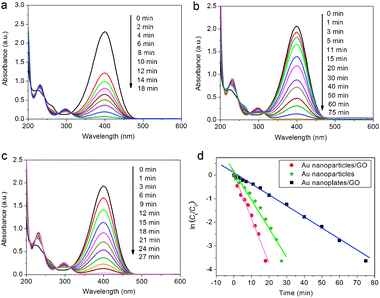 | ||
| Fig. 3 UV-vis adsorption spectra of the reduction of 4-NP by NaBH4 in the presence of Au nanoparticles/GO (a), Au nanoplates/GO (b), and Au nanoparticles alone (c); (d) plot of ln(Ct/C0) of 4-NP against time for the catalysts. | ||
In summary, we have developed a relatively green and environment friendly route for the synthesis of Au nanostructures with controllable morphologies on TA-functionalized GO using TA as a reducing and stabilizing agent. These Au nanostructures/GO nanocomposites are found to exhibit good catalytic performance toward 4-NP reduction. The GO supports not only enhance the catalytic activity of Au nanostructuresvia a synergistic effect, but also lend the Au nanostructures/GO nanocomposites the easiness of separation and recovery for practical catalytic applications.
Acknowledgements
This work was supported by National Basic Research Program of China (No. 2011CB935800).Notes and references
- (a) Y. Sun and Y. Xia, Science, 2002, 298, 2176 CrossRef CAS; (b) B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709 CrossRef CAS; (c) M. Schrinner, M. Ballauff, Y. Talmon, Y. Kauffmann, J. Thun, M. Möller and J. Breu, Science, 2009, 323, 617 CrossRef CAS; (d) M. D. Hughes, Y. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132 CrossRef CAS; (e) J. Zeng, Q. Zhang, J. Chen and Y. Xia, Nano Lett., 2010, 10, 30 CrossRef CAS; (f) C. Richard, M. M. Q. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181 CrossRef CAS.
- (a) S. E. Skrabalak, J. Y. Chen, Y. G. Sun, X. M. Lu, L. Au, C. M. Cobley and Y. Xia, Acc. Chem. Res., 2008, 41, 1587 CrossRef CAS; (b) B. Wiley, Y. G. Sun and Y. Xia, Acc. Chem. Res., 2007, 40, 1067 CrossRef CAS; (c) J. Schulz, A. Roucoux and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS.
- (a) S. Saha, A. Pal, S. Kundu, S. Basu and T. Pal, Langmuir, 2010, 26, 2885 CrossRef CAS; (b) X. Hu, T. Wang, X. Qu and S. Dong, J. Phys. Chem. B, 2006, 110, 853 CrossRef CAS; (c) S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814 CrossRef CAS; (d) K. Y. Lee, Y. W. Lee, K. Kwon, J. Heo, J. Kim and S. W. Han, Chem. Phys. Lett., 2008, 453, 77 CrossRef CAS; (e) Y. Chang and D. Chen, J. Hazard. Mater., 2009, 165, 664 CrossRef CAS.
- (a) H. Koga, E. Tokunaga, M. Hidaka, Y. Umemura, T. Saito, A. Isogai and T. Kitaoka, Chem. Commun., 2010, 46, 8567 RSC; (b) S. Tang, S. Vongehr and X. Meng, J. Mater. Chem., 2010, 20, 5436 RSC; (c) K. Hayakawa, T. Yoshimura and K. Esumi, Langmuir, 2003, 19, 5517 CrossRef CAS; (d) J. Zhang, J. Lei, R. Pan, C. Leng, Z. Hu and H. Ju, Chem. Commun., 2011, 47, 668 RSC; (e) L. Ouyang, D. M. Dotzauer, S. R. Hogg, J. Macanás, J. Lahitte and M. L. Bruening, Catal. Today, 2010, 156, 100 CrossRef CAS; (f) H. Jiang, B. Liu, T. Akita, M. Haruta, H. Sakurai and Q. Xu, J. Am. Chem. Soc., 2009, 131, 11302 CrossRef CAS; (g) X. Liu, A. Wang, X. Wang, C. Mou and T. Zhang, Chem. Commun., 2008, 3187 RSC; (h) H. Jiang, T. Umegaki, T. Akita, X. Zhang, M. Haruta and Q. Xu, Chem.–Eur. J., 2010, 16, 3132 CrossRef CAS; (i) Z. Y. Zhang, C. L. Shao, P. Zou, P. Zhang, M. Y. Zhang, J. B. Mu, Z. C. Guo, X. H. Li, C. H. Wang and Y. C. Liu, Chem. Commun., 2011, 47, 3906 RSC.
- (a) X. Huang, S. Z. Li, Y. Z. Huang, S. X. Wu, X. Z. Zhou, S. Z. Li, C. L. Gan, F. Boey, C. A. Mirkin and H. Zhang, Nat. Commun., 2011, 2, 292 Search PubMed; (b) X. Huang, X. Z. Zhou, S. X. Wu, Y. Y. Wei, X. Y. Qi, J. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 513 CrossRef CAS; (c) X. Huang, Z. Y. Yin, S. X. Wu, X. Y. Qi, Q. Y. He, Q. C. Zhang, Q. Y. Yan, F. Boey and H. Zhang, Small, 2011 DOI:10.1002/smll.201002009; (d) X. Z. Zhou, X. Huang, X. Y. Qi, S. X. Wu, C. Xue, F. Y. C. Boey, Q. Y. Yan, P. Chen and H. Zhang, J. Phys. Chem. C, 2009, 113, 10842 CrossRef CAS; (e) B. S. Kong, J. Geng and H. T. Jung, Chem. Commun., 2009, 2174 RSC.
- J. Huang, L. M. Zhang, B. Chen, N. Ji, F. H. Chen, Y. Zhang and Z. J. Zhang, Nanoscale, 2010, 2, 2733 RSC.
- Y. D. Lei, Z. H. Tang, R. J. Liao and B. C. Guo, Green Chem., 2011, 13, 1655 RSC.
- (a) X. Huang, Y. P. Wang, X. P. Liao and B. Shi, Chem. Commun., 2009, 4687 RSC; (b) X. Huang, H. Wu, X. P. Liao and B. Shi, Green Chem., 2010, 12, 395 RSC.
- (a) Y. Du, H. L. Chen, R. Z. Chen and N. P. Xu, Appl. Catal., A, 2004, 277, 259 CrossRef CAS; (b) J. F. Corbett, Dyes Pigm., 1999, 41, 127 CrossRef CAS; (c) C. V. Rode, M. J. Vaidya and R. V. Chaudhari, Org. Process Res. Dev., 1999, 3, 465 Search PubMed; (d) Z. Y. Zhang, C. L. Shao, P. Zou, P. Zhang, M. Y. Zhang, J. B. Mu, Z. C. Guo, X. H. Li, C. H. Wang and Y. C. Liu, Chem. Commun., 2011, 47, 3906 RSC.
- (a) Y. Deng, Y. Cai, Z. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2010, 132, 8466 CrossRef CAS; (b) H. Jiang, T. Akita, T. Ishida, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2011, 133, 1304 CrossRef CAS; (c) M. Yu, S. Geeta, L. Yan and B. Matthias, Langmuir, 2005, 21, 12229 CrossRef CAS.
- S. Praharaj, S. Nath, S. K. Ghosh, S. Kundu and T. Pal, Langmuir, 2004, 20, 9889 CrossRef CAS.
- (a) S. Tang, S. Vongehr and X. Meng, J. Phys. Chem. C, 2010, 114, 977 CrossRef CAS; (b) Y. Mei, Y. Lu, F. Polzer and M. Ballauff, Chem. Mater., 2007, 19, 1062 CrossRef CAS.
- J. F. Huang, S. Vongehr, S. C. Tang, H. M. Lu and X. K. Meng, J. Phys. Chem. C, 2010, 114, 15005 CAS.
- R. Leary and A. Westwood, Carbon, 2011, 49, 741 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary figures. See DOI: 10.1039/c1cy00205h |
| This journal is © The Royal Society of Chemistry 2011 |