Catalytic properties of cobalt and nickel ferrites dispersed in mesoporous silicon oxide for ethylbenzene dehydrogenation with CO2
Tiago Pinheiro
Braga
a,
Bárbara Maria Campos
Sales
a,
Antonio Narcisio
Pinheiro
a,
W. T.
Herrera
b,
E.
Baggio-Saitovitch
b and
Antoninho
Valentini
*a
aLangmuir - Laboratório de Adsorção e Catálise, Departamento de Química Analítica e Físico-Química, Universidade Federal do Ceará, Campus do Pici, CEP 60455-970, Fortaleza CE, Brasil. E-mail: valent@ufc.br; Fax: +55 85 3366 9982; Tel: +55 85 3366 9951
bDepartamento de Física Experimental, Centro Brasileiro de Pesquisas em Física, Rua Dr. Xavier Sigaud 150, CEP 22290-180, Rio de Janeiro RJ, Brasil
First published on 6th September 2011
Abstract
A polymeric precursor method was applied to synthesize catalysts of the general formula MFe2O4 (M = Co and Ni) in order to contribute to studies on ethylbenzene dehydrogenation in the presence of CO2. The catalysts were characterized by TG, H2-TPR, XRD, Mossbauer spectroscopy (MS), N2 adsorption/desorption isotherms and TPD-CO2. Investigations using XRD and MS revealed that the spinel structures of CoFe2O4 and NiFe2O4 phases were formed. The catalytic results suggested that materials with a spinel structure are particularly interesting for ethylbenzene dehydrogenation. Compared to the other catalysts synthesized the sample containing cobalt ferrite showed higher conversion and good styrene selectivity. The analysis of the spent catalyst (DRX) showed that the CoFe2O4 phase was stable under the reaction conditions and that a coke (TG) deposit was more pronounced for the NiFeSi.
1. Introduction
Catalytic ethylbenzene dehydrogenation in the presence of steam (reaction (1)) is a representative process for the production of styrene, an important monomer for synthetic polymers.1–4C6H5–CH2CH3(g) ⇌ C6H5–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2(g) + H2(g) CH2(g) + H2(g) | (1) |
Currently, researchers have studied the use of carbon dioxide (CO2) as a co-feeder gas in ethylbenzene dehydrogenization (reaction (2)) with the aim of introducing an alternative route to the steam process.7–10
| C6H5–CH2CH3(g) + CO2(g) ⇌ C6H5–CHCH2(g) + CO(g) + H2O(g) | (2) |
Various publications have shown that, for catalysts containing hematite as the active phase, magnetite forms during the dehydrogenation reaction, which subsequently affects the conversion of ethylbenzene to styrene.4,5,9,12 On the other hand, there are some publications pointing to materials with spinel structures, such as ferrites, as presenting remarkable catalytic activity and stability in this reaction.13,14 In addition to their attractive redox and acid/base properties, spinel structures are versatile compounds offering structural stability.15 Therefore, materials of the general type MFe2O4 can be used in heterogeneous catalysis.15–18 Specifically in the ethylbenzene dehydrogenization reaction, the ferrites of Co and/or Ni may be interesting due to their dehydrogenation ability. Additionally, these ions (Co2+ or Ni2+) may occupy different positions in the spinel structure, changing the interactions of CO2 with the catalyst surface and the redox properties of the Fe3+, which is the main site for ethylbenzene adsorption.19
The synthesis of ferrites of Co and Ni with nanometric size has already been described in the literature.20–22 Nevertheless, in order to apply these materials (ferrites of Co and Ni with nanometric size) in heterogeneous catalysis, it is necessary to avoid the sintering process. In this context, we report the synthesis and characterization of MFe2O4 (M = Ni or Co) compounds embedded in SiO2 with the purpose of studying their catalytic performance for ethylbenzene dehydrogenation in the presence of carbon dioxide.
2. Experimental
2.1. Catalyst preparation
The precursor polymeric method used in this report was based on the chelation of cations (metals) by citric acid in aqueous solution.23–25Nickel nitrate hexahydrate {Ni(NO3)2·6H2O}, cobalt nitrate hexahydrate {Co(NO3)2·6H2O}, iron nitrate nonahydrate {Fe(NO3)3·9H2O}, tetraethylorthosilicate (TEOS), citric acid monohydrate (CA){C6H8O7·H2O} and ethylene glycol (EG) {C2H6O2} were used as starting materials. A CA/metal ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (mol) was used for all samples; the metal amount was the sum of Fe3+, Co2+ or Ni2+ and Si4+, and the mass ratio of CA/EG was kept at 2/3. The different samples were denoted by the general formula MFeSi, where M was the transition metal (Co or Ni) used. In addition, a sample containing only iron oxide and silicon oxide was prepared and labeled FeSi.
:1 (mol) was used for all samples; the metal amount was the sum of Fe3+, Co2+ or Ni2+ and Si4+, and the mass ratio of CA/EG was kept at 2/3. The different samples were denoted by the general formula MFeSi, where M was the transition metal (Co or Ni) used. In addition, a sample containing only iron oxide and silicon oxide was prepared and labeled FeSi.
For the synthesis of the CoFeSi sample, 0.0085 mol and 0.017 mol of Co and Fe precursors, respectively, were dissolved in distilled water at room temperature. CA (0.151 mol) was dissolved in ethanol at 50 °C followed by the addition of 0.05 mol of a Si precursor (TEOS) with stirring. Next, the aqueous solution was added to the CA-TEOS-ethanol solution and stirred for 15 min at 50 °C. Subsequently, the EG was added and the mixture was stirred at 100 °C until it became a viscous resin. The resin was treated at 300 °C for 1 h under air, and the resulting precursor composite was milled and treated at 700 °C under a flow of air for 1 h.
2.2. Catalyst characterization
The calcination step was followed by thermogravimetric analysis (TG) using a 10 °C min−1 heating rate under a flow of air at 50 ml min−1 for 10 mg of a sample. The crystal structure of the metal oxides was characterized by X-ray diffraction analysis (XRD) with a CuKα irradiation source (λ = 1.5418 Å, 30 kV and 20 mA). Transmission Mössbauer spectra were recorded at room temperature and at 4.2 K in constant acceleration mode with a 57Co (Rh) source. The Mössbauer data were fitted to discrete Lorentzian functions using the least-square fitting routine of the NORMOS® software package. All isomer shift values (δ) were quoted relative to αFe.Specific surface area and pore volume of the catalysts were determined by N2 adsorption/desorption isotherms at liquid nitrogen temperature. Before N2 adsorption, the catalyst sample (40 to 50 mg) was evacuated at 200 °C for 2 h. The TPR analyses (temperature-programmed reduction) of the catalysts were carried out in the range of 100–950 °C in a quartz reactor by passing a 5% H2/N2 flow (30 ml min−1) over the sample at a heating rate of 10 °C min−1. The H2 consumption was monitored by a thermal conductivity detector (TCD) connected to the reactor outlet. The CO2 temperature-programmed desorption was carried out in the range of 40–500 °C under a He flow (10 °C min−1, 30 ml min−1). The catalysts (200 mg) were preheated under He flow (30 ml min−1) at 550 °C for 1 h, after which the temperature was decreased to 60 °C and the He flow was changed to pure CO2 (30 ml min−1 for 0.5 h). The desorbed CO2 was detected by an online thermal conductivity detector (TCD) after passing through a trap (−20 °C) to remove any trace of water.
To assess the amount and the properties of coke deposited, the spent catalysts were analyzed by thermogravimetric analysis (TG) using a 10 °C min−1 heating rate under a flow of air at 50 ml min−1 and Fourier Transform Infrared (FTIR) in the range of 400 to 4000 cm−1 using KBr pellets containing 1.0 wt% of the sample.
2.3. Catalytic activity
Catalytic tests were carried out in a fixed bed stainless steel microreactor using 150 mg of a sample at 550 °C and atmospheric pressure with a CO2 to ethylbenzene molar ratio of 30. The molar flow rate of ethylbenzene was controlled at 1.446 mmol h−1. Prior to reaction, the catalyst was heated under a N2 flow (30 ml min−1) up to the reaction temperature, and the reaction mixture (CO2, ethylbenzene and N2) was then introduced at a flow rate of 30 ml min−1. The resulting catalytic ethylbenzene conversion was analyzed by gas chromatography using an instrument equipped with a FID and a nonpolar capillary column (30 m × 0.25 mm × 0.25 μm, similar to AT-1). The ethylbenzene conversion (CEt) was calculated according to eqn (1), and the styrene selectivity (SSt) was calculated according to eqn (2).| CEt = (ethylbenzenein − ethylbenzeneout)/ethylbenzenein | (1) |
| SSt = (styreneout)/n(hydrocarbon products) | (2) |
3. Results and discussion
3.1. Thermal analysis
As in previous publications,23–25 the TG/DTG curves (not shown) could be divided into two temperature ranges for weight loss. The first range (40–300 °C) (near 10 wt%) was related to the evaporation of adsorbed water and any remaining ethylene glycol. The second stage (300–450 °C) was related to the decomposition of the residual organic material of the polymeric chains formed by polyesterification25,26 and subsequent production of CO2 by the decomposition of residual organic precursors (near 50 wt%). After that, no weight loss was detected up to 700 °C, which suggested that the decomposition of organic material had finished.Employing organic precursors (CA and EG), which were removed as volatile compounds during the calcination step in the preparation of the catalysts, resulted in cavities and the simultaneous rearrangement of the solid, which might have been responsible for the morphological properties displayed by the material (pore diameter and volume).
3.2. X-Ray diffraction
After the calcination process (700 °C under airflow), the catalyst powders were analyzed by X-ray diffraction (XRD) to gather information on the solid phase formed (Fig. 1). In spite of the low crystalline organization, the XRD patterns of the catalyst CoFeSi suggested the formation of a spinel structured phase (CoFe2O4, JCPDS 02-1045). Likewise, the sample NiFeSi also pointed to the spinel structure of nickel ferrite (NiFe2O4, JCPDS 44-1485) in addition to the presence of nickel oxide (NiO, JCPDS 78-0423). The FeSi sample showed a hematite pattern (α-Fe2O3, JCPDS 86-2368) and a maghemite phase (γ-Fe2O3, JCPDS 39-1346). | ||
| Fig. 1 X-Ray diffraction profile of the catalysts after the calcination process. | ||
Due to the similarity in the diffraction profile of the nickel ferrite and cobalt ferrite with the maghemite phase (γ-Fe2O3), the presence of a γ-Fe2O3 phase could not be initially ruled out for CoFeSi and NiFeSi samples.
From the results shown above, it seemed that nickel ferrite (NiFe2O4) was easier to form than cobalt ferrite (CoFe2O4) despite the fact that the ionic radii of Fe3+ (0.67 Å) were smaller than the radii of Ni2+ (0.83 Å) and closer to Co2+ (0.68 Å). It therefore seemed reasonable to assume that a replacement of the Fe3+ ions by the Co2+ ions in both the tetrahedral A and octahedral B sites of the cation sublattices with the corresponding decrease in the lattice parameters27 would be more favourable than by the larger Ni2+ ions.
To clarify the XRD results, the diagrams were analyzed by means of the Rietveld refinement procedure28 from which the crystallographic lattice parameters of the samples were determined. To achieve this, the DBWS9807 program described by Young et al.29 was applied. By this method it was possible to quantify the different phases formed and the resulting data are listed in Table 1. The crystallite sizes of the samples were calculated from the XRD patterns using the Debye–Scherrer formula.30 The calculated results illustrated in Table 1 indicated that the catalysts were nanometre-sized crystalline powders and probably with a high number of crystalline defects.
3.3. Mössbauer spectroscopy
As observed in the XRD results, the small size of the particles synthesized and the slight difference in the lattice parameters of the maghemita (γ-Fe2O3) and ferrite phases (CoFe2O4 and NiFe2O4) made the XRD insufficient for characterization of the powders. As shown in Fig. 2a, there were two sextets very close to one another and a superparamagnetic central doublet in the Mössbauer spectra of FeSi at room temperature. In addition, one six-line subspectra with an isomer shift of δ = 0.30 mm s−1 relative to αFe, a quadrupole shift of ε = −0.20 mm s−1, and a magnetic hyperfine field Bhf = 51.5 tesla were also observed. This type of pattern is typical of hematite (α-Fe2O3). The other one six-line subspectra had δ = 0.50 mm s−1, a quadrupole shift of ε = ∼0.02 mm s−1, and Bhf = 50 tesla, which are all characteristics of maghemite.31 | ||
| Fig. 2 57Fe Mössbauer spectra of the ferrites, at room temperature (300 K) and at 4.2 K. FeSi (a), CoFeSi (b) and NiFeSi (c). | ||
Alternatively, the Mössbauer spectrum of the FeSi sample was also acquired at a low temperature because the influence of superparamagnetism could be counteracted by reducing the sample temperature. The superparamagnetic relaxation was suppressed at 4.2 K (Fig. 2a), but the presence of different iron environments in the sample was evidenced by two six-line subspectra. This result corroborated with XRD observations on the presence of hematite and maghemite phases in the samples.
Similarly, as shown in Fig. 2b and c, the room temperature Mössbauer spectra of the CoFeSi and NiFeSi samples exhibited the superposition of a magnetic split sextet pattern and a superparamagnetic doublet. The Mössbauer spectrum at 4.2 K for the sample CoFeSi was fitted with four subspectra. Two six-line subspectra with isomer shifts of δ = 0.25 and 0.38 mm s−1 (relative to αFe), quadrupole shifts of ε = 0.02 and −0.08 mm s−1, and magnetic hyperfine fields of Bhf = 50.3 and 52.3 tesla were attributed to cobalt ferrite. These hyperfine parameters corresponded to a distribution of Fe in site A (tetrahedral) and site B (octahedral). Nevertheless, a broad sextet with Bhf = 30–49 tesla was also observed, which was probably due to cobalt ferrite with an extremely small particle size. The broad sextet is typical for Fe particles in the magnetic relaxation regime. Moreover, at 4.2 K, great deals of magnetic moments were blocked, as evidenced by the appearance of a broad and asymmetric magnetic sextet.32 In Fig. 2b (at 4.2 K), we see that 90% of particles were blocked and the remaining central doublet (10%) suggested that there were very small superparamagnetic particles (less than 4 nm). These results were in agreement with the mean diameter for a CoFeSi sample (4 nm), as estimated by XRD analysis (Table 1).
The Mössbauer spectrum at 4.2 K for the NiFeSi sample was fitted with three magnetic subspectra. The hyperfine field values for sites A (tetrahedral) and B (octahedral) were 49.6 and 52.8 tesla, respectively. The corresponding isomer shift values were 0.31 and 0.40 mm s−1 relative to αFe. In addition, one broad sextet with Bhf = 30–51 tesla was observed, which was probably due to nickel ferrite with a very small particle size. In addition, the decrease of the magnetic hyperfine field from the bulk value could be attributed to the very small particle size.33 In the spectra of NiFeSi (Fig. 2c, 4.2 K), unlike CoFeSi (Fig. 2b, 4.2 K), we saw that 100% of the particles were blocked, suggesting that the size of the nanoparticles was larger than 4 nm of diameter. Thus, the Mössbauer results were in agreement with the X-ray diffraction patterns, which suggested a particle diameter of 11.4 nm (Table 1).
3.4. Temperature-programmed reduction
To acquire more information on the properties of the catalysts and to clarify the chemical environment of the iron oxide, which influences the reducibility of the catalyst, temperature-programmed reduction (TPR) analyses were carried out (Fig. 3). At first the TPR pattern of the FeSi catalyst suggested four peaks of reduction. However, this pattern is somewhat complex and was decomposed into five well-defined Gaussian-shaped peaks and an incomplete one at high temperature (agreement factor of 0.997). | ||
| Fig. 3 Temperature-programmed reduction (TPR) profiles of the samples. | ||
The H2 consumption attained a maximum at 430 °C and was related to the reduction of Fe3+ to Fe2+, probably due to the formation of the magnetite phase (Fe3O4).9,12 In addition, the peak at 575 °C, which overlapped with the first (430 °C) and the other peaks at higher temperatures, might have been due to the reduction of Fe2+ to Fe0. This low reduction temperature (575 °C) might have been the result of the small particle diameter, as indicated by XRD analysis (Table 1). Furthermore, the H2 consumption peak at 575 °C was probably due to Fe2+ reduction because of the amount of H2 consumption observed for the peaks at higher temperatures, which was lower than the peak at 430 °C. This phenomenon suggested either incomplete Fe2+ reduction or the formation of FeO instead of the desired Fe3O4 phase at 430 °C. Furthermore, certainly the three H2 consumption peaks in the temperature range of 600–820 °C (with a maximum at 645, 705 and 780 °C), as well as a peak at temperatures higher than 900 °C, were associated with Fe2+ reduction to produce metallic iron.12,34
A possible explanation for the presence of four H2 consumption peaks at temperature ranges higher than 600 °C for the FeSi sample might be drawn from the textural properties. Metal oxide particles placed inside of porous materials are more difficult to reduce compared to metal oxides positioned on external surfaces.12,35 Therefore, the peak at 645 °C might have been due to Fe2+ reduction outside of the porous channel, while the peaks at 705 and 780 °C and higher than 900 °C might have been from the reduction of Fe2+ inside the porous channel. Furthermore, the TPR profile of the FeSi sample suggested (as did the XRD) a high proportion of iron in the Fe3+ oxidation state, which was interesting for dehydrogenation.4,9,12
The TPR profile of the NiFeSi catalyst had a starting H2 consumption process near 200 °C, and a shoulder at 520 °C was clearly observed. The pattern was decomposed into five well-defined Gaussian-shaped peaks and an incomplete one at high temperature (agreement factor of 0.998). The peak at 414 °C could be attributed to the Ni2+ and Fe3+ (to Fe2+) reduction processes. Similar to the FeSi sample, the peak at 426 °C could be attributed to the reduction of Fe3+ to Fe2+. The Ni2+ reduction in a temperature range was typical for a nickel oxide phase dispersed in SiO2,36 which was determined to be present by XRD analysis (Fig. 1). Nevertheless, the peak at 500 °C could be attributed to the reduction of Ni2+ in the spinel phase, which had an intense interaction with the iron oxide and might have been localized in octahedral or tetrahedral positions. The peaks at temperatures higher than 600 °C were assumed to be the result of Fe2+ reduction.
The TPR profile of the CoFeSi catalyst presented a poorly defined pattern, but it was successfully decomposed into seven well-defined Gaussian-shaped peaks and an incomplete one at high temperature (agreement factor of 0.998). The weak peak at 345 °C was associated to the reduction of the Co3O4 phase (Co3+ to Co2+). The low H2 consumption observed corroborated with the XRD analysis, which did not indicate the presence of this phase. The presence of this phase might have been missed, however, due to a smaller proportion. The peak at 436 °C was attributed mainly to the reduction of Fe3+ (to Fe2+). Similar to the NiFeSi sample, the peaks at 518 °C and at 524 °C could be also attributed to the reduction of Fe3+ to Fe2+ and the reduction of Co2+ to Co0, both in the spinel phase and possibly localized in octahedral or tetrahedral positions. The peaks for temperatures higher than 600 °C might have been due to Fe2+ reduction influenced by the cobalt presence and/or textural properties.
Different from the NiFeSi sample the CoFeSi catalyst showed low H2 consumption peaks in the temperature range of 600 °C to 700 °C. In addition, it is observed a well defined H2 consumption peak at 840 °C, which may be due to the CoFe2O4 phase reduction.
The H2 consumption peaks that appeared at temperatures higher than 600 °C were particularly interesting for ethylbenzene dehydrogenation because they provided evidence that the reaction temperature (550 °C) did not completely reduce the transitions metals.
Assuming that the iron oxide species (Fe3+) was the main active site for ethylbenzene dehydrogenation,6 the TPR profiles suggested that the addition of nickel additives could not delay the deactivation process during the catalytic test. However, and at the same level of importance, the TPR patterns pointed to a mixture of different oxide phases, as did XRD and Mössbauer analysis. In addition to the low particle diameter (Table 1), this heterogeneity of phases in the catalyst composition suggested that these samples might present a significant number of defects in the surface, an interesting phenomenon for the catalytic process.
On the other hand, the H2 consumption profile in a temperature range higher than 600 °C for the CoFeSi sample suggests that cobalt addition could delay the catalyst deactivation due to a reduction process during the catalytic test carried out at 550 °C.
3.5. Specific surface area measurements
From the perspective of the catalysis and the catalytic characterization, the surface area determination was an important parameter for application. Thus, the N2 adsorption/desorption isotherms were measured and the isotherm profile and pore diameter distribution are presented in Fig. 4a and b. The samples CoFeSi and NiFeSi showed similar isotherm profiles that were characteristic of mesoporous materials (type IV according to IUPAC classification and the H2 hysteresis loop). This behavior indicated that the pore structure was complex and consisted of interconnected networks of pores of different sizes and shapes. On the other hand, the sample FeSi showed isotherms of type II characteristics of mesoporous powders (H3 hysteresis loop). A type H3 loop is usually assigned to slit-shaped pores due to non-rigid aggregates of particles.37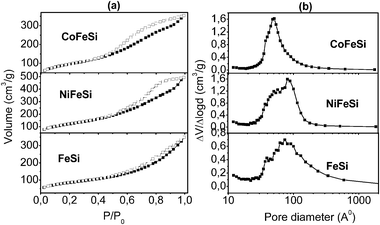 | ||
| Fig. 4 N2 adsorption/desorption isotherms of the catalysts (a). Pore diameter distribution of the samples (b) (calculated using the BJH model). | ||
The profiles of pore diameter distribution are presented in Fig. 4b (according to the BJH method) and exhibited a narrow pore size distribution, with the majority of the diameters located in the range of 16 to 238 Å. Additionally, Table 2 lists the surface area, total pore volume and average pore diameter of the studied powders. In spite of the similarity in the average pore diameter and pore diameter distribution (Fig. 4b) for the samples, the addition of transition metals (Co and Ni) to the catalyst promoted the synthesis of samples with better textural properties. For instance, the catalyst NiFeSi possessed higher specific surface area (49% higher) and total pore volume (40% higher) in comparison with the FeSi sample, while the sample CoFeSi showed a specific surface area 18% higher than the FeSi sample.
| Sample | S/m2 g−1 | V p/cm3 g−1 | D p/Å |
|---|---|---|---|
| CoFeSi | 346 | 0.55 | 63 |
| NiFeSi | 439 | 0.76 | 70 |
| FeSi | 294 | 0.54 | 74 |
Such textural effects in the porosity of the synthesized solids might be related to the ratio of residual organic precursors, as verified in the TG analysis. As such, samples with a spinel structure (CoFeSi and NiFeSi) were seen to display a higher ratio of residual organic substances. After calcination, this can result in differences in the characteristics of the pores, which are perceptible in the values for the surface area, pore volume, pore diameter and changes in the hysteresis behaviour.
3.6. Temperature-programmed desorption
It is accepted that the basic sites play an important role in ethylbenzene dehydrogenation.19 Therefore, it was interesting to determine the effect of nickel and cobalt addition on the basicity of the catalyst samples. To verify the relative basicity, temperature-programmed desorption of CO2 (TPD-CO2) experiments was carried out (Fig. 5). The data in Fig. 5 are in arbitrary units, and the intensity of the deflection was not quantified with regard to the number of moles of CO2. Nevertheless, the experimental conditions were rigorously the same for all samples, and thus it was possible to compare the areas of the TPD profiles. | ||
| Fig. 5 Temperature-programmed desorption of CO2 (TPD-CO2) for the catalysts after calcination. | ||
The TPD patterns of NiFeSi and FeSi samples showed one peak for CO2 desorption, while the catalyst CoFeSi presented two peaks for desorption. The maximum desorption peak for NiFeSi was observed around 90 °C, whereas the maximum for FeSi was around 118 °C. On the other hand, the desorption curves corresponding to the CoFeSi sample were around 97 and 368 °C, respectively. The patterns of TPD-CO2 were decomposed for all samples into two or three well-defined Gaussian-shaped peaks. Therefore, different types of basic sites (or CO2 to metal oxides interactions) might be distinguished.
The first CO2 desorption temperature range (53–200 °C) was attributed to weak basicity. However, the second CO2 desorption temperature range (337–420 °C) was typical of a medium basicity. Thus, the peak of CO2 desorption at 370 °C suggested that the CoFeSi sample had a higher basic site strength. Consequently, it was expected that the CoFeSi would have the highest ethylbenzene conversion, followed by FeSi and then NiFeSi. Considering the amount of CO2 desorbed from the sample (the total areas of the TPD profiles), the number of basic sites followed the order: FeSi > CoFeSi > NiFeSi. NiFeSi stood out as a solid with relatively low basicity, while FeSi had a large amount of basic sites.
In general, basic catalysts can improve the adsorption of acidic CO2 to supply the surface with oxygen, suppressing the deposition of coke.38 Additionally, the number and strength of the basic sites of the catalyst had an effect on the catalytic performance.39 Several research papers12,40 have shown that the catalytic activity for ethylbenzene dehydrogenization is increased with improved CO2 adsorption ability.
3.7. Catalytic performance
The ethylbenzene conversion and styrene selectivity for the catalytic samples are depicted in Fig. 6a and b. All the samples presented a similar activity and showed a reasonable selectivity in ethylbenzene dehydrogenization. However, some important differences were observed.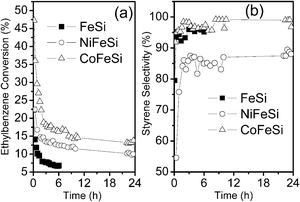 | ||
| Fig. 6 Catalytic performance as a function of time for the catalysts: ethylbenzene conversion (a), styrene selectivity (b). | ||
The CoFeSi sample demonstrated a higher ethylbenzene conversion and styrene selectivity than either FeSi or NiFeSi catalyst. Initially, the NiFeSi and FeSi catalysts presented a similar ethylbenzene conversion (near 25%); however, the catalytic deactivation was higher for the FeSi sample.
Analysis of the byproducts observed for the samples can help in the interpretation of catalyst properties. Considering benzene selectivity (Table 3), which is the main byproduct for all the catalysts tested, it is important to point out that the NiFeSi sample showed the highest benzene selectivity (near 23%) up to 120 min. At first, a superior benzene production meant higher catalyst acidity and consequently, more significant carbon deposition. Taking into account the TPD-CO2 measurements, the NiFeSi sample presented the lowest ability for CO2 adsorption by mass of any catalyst tested, which meant higher acidity relative to the FeSi and CoFeSi samples.
The deactivation of catalysts may occur due to the reduction of the active phase and/or by blocking of the surface by carbonaceous deposits.12 To investigate the cause of catalyst deactivation during ethylbenzene dehydrogenization, studies were conducted to verify possible changes in the chemical properties of the catalysts, as well as determining the amount and nature of coke deposits on the catalysts. For this, XRD analysis after the catalytic tests were performed, and the results are depicted in Fig. 7. These data made it possible to verify some differences from the fresh catalysts (Fig. 2).
 | ||
| Fig. 7 X-Ray diffraction profiles of the samples after the catalytic tests. | ||
The pattern presented in Fig. 7 for the FeSi sample pointed to Fe3+ reduction during the reaction process. In addition, the diffraction pattern of the FeSi sample pointed to magnetite phase formation (Fe3O4 JCPDS 26-1136) and the maghemite phase was still detected (γ-Fe2O3 JCPDS 39-1346).
Through careful XRD pattern (Fig. 7) analysis, refinement based on the Rietveld method28 was carried out, and the weight percentages for the different phases detected are listed in Table 4. Interestingly, for the FeSi sample, maghemite was the major phase (89 wt%) in the used sample but not the fresh sample (Table 1), suggesting that the maghemite phase was formed from hematite during the reaction process simultaneously to magnetite phase formation. From the literature,41 however, the hematite phase has a superior stability to maghemite. In spite of the fact that experimental data presented here are not enough to support this model, a possible explanation for the observed result was the formation of maghemite particles within a shell of hematite. These core–shell particles were then partially reduced during ethylbenzene dehydrogenation, maintaining the maghemite in the interior. If Fe2+ was not as active in ethylbenzene dehydrogenation as Fe3+,13 the core–shell particle formation theory might explain the observed decreases in the conversion of ethylbenzene presented in Fig. 6a.
| Crystalline phase (%) | Samples | ||
|---|---|---|---|
| CoFeSi | NiFeSi | FeSi | |
| NiFe2O4 | — | 27 | — |
| CoFe2O4 | 100 | — | — |
| γ-Fe2O3 | — | — | 89 |
| Fe3O4 | — | 11 | |
| NiO | — | 64 | — |
| Ni | — | 9 | — |
On the other hand, it is necessary to carry out analysis in order to clarify if the reaction conditions (CO2 presence) are favourable to maghemite formation.
The reaction process also promoted the partial reduction of the NiFeSi sample, forming metallic nickel (Ni, JCPDS 01-1258). However, the NiFeSi pattern suggested that a ratio of the structures of nickel ferrite (NiFe2O4, JCPDS 44-1485) and nickel oxide (NiO, JCPDS 78-0423) was maintained after 24 h of reaction time. On the other hand, the relative mass percentage (Table 4) calculated indicated a decrease in the wt% of NiFe2O4 and an increase for NiO. These results suggested that the Fe3+ from the spinel phase was reduced, which then likely promoted magnetite phase formation without Ni2+ reduction. This hypothesis was suggested because the NiO wt% for the used sample (Table 4) was higher than for the fresh sample (Table 1). This observation might be related to a superior stability for the Ni2+ species relative to the Fe3+ or that a higher proportion of tetrahedral sites were occupied by Ni2+. The disproportionate occupation would consequently lead to less exposure to the reduction process. The metallic nickel formed might have been from the reduction of the NiO detected in the fresh sample. Furthermore, it is well known that nickel in the metallic state (Ni0) is able to promote a C–H bond disproportionation reaction,42 which for some reactions is responsible for a high ratio of coke deposition.13,42 Thus, it was expected that the NiFeSi sample would present a high amount of coke deposition.
In contrast to the other samples, the CoFeSi catalyst seemed to not undergo phase modification due to the reaction process. The XRD pattern of CoFeSi after the reaction was very similar to that for the fresh catalyst (Fig. 1), which indicated a superior stability for the CoFe2O4 phase under the reaction conditions. If the change did occur, it was in a very low proportion. In addition, taking into account the by-product selectivity (Table 3), the CoFeSi sample showed a lower benzene selectivity, which suggested a low coke deposition.
Therefore, the XRD results strengthen the suggestion that the H2 consumption peak at 840 °C showed by the CoFeSi sample (Fig. 3) may be due to CoFe2O4 phase reduction.
In spite of the valuable information acquired from the catalytic test expressed in Fig. 6, the reaction time used for the FeSi sample was only of 6 h due to the high deactivation. Therefore, catalytic tests with a period of 24 h were carried out for the samples CoFeSi and NiFeSi. The respective ethylbenzene conversions were 13% and 10%, which suggested the same rate of deactivation for both samples over 24 h. Additionally, the ethylbenzene conversion observed for the NiFeSi suggested that high surface area alone was not a determining factor for superior catalytic conversion. In spite of the higher surface area presented by the NiFeSi catalyst (Table 2), the ethylbenzene conversion values were lower than the CoFeSi sample. Therefore, the CoFeSi catalyst was the most active solid (conversion near 17% after 4 h) due to the combined effects of basicity, surface area and reducibility. Despite the H2-TPR profile of CoFeSi resembling that of NiFeSi, the XRD analysis of the spent catalysts revealed that the spinel structure of the CoFeSi sample was maintained, presenting a hard reducibility under the ethylbenzene dehydrogenation conditions and the temperature-programmed desorption of CO2 revealed a medium basicity for the CoFeSi sample, justifications for its better catalytic performance.
To find another reason for the decrease in ethylbenzene conversion, the spent catalysts were analyzed by thermogravimetric analysis (TG) (Fig. 8).
 | ||
| Fig. 8 Thermogravimetric profile of the samples carried out under synthetic air flow. NiFeSi and CoFeSi after the catalytic test of 24 h, FeSi after 6 h of time on stream. | ||
This thermogravimetric analysis of the spent catalysts was carried out to estimate the amount of coke deposited, as revealed by the reduction in weight due to coke oxidation in air. Notice that the thermal analysis results (Fig. 8) pointed to a lower coke deposition for FeSi, while the NiFeSi catalyst presented a higher coke deposition. The thermograms of three coked catalyst samples revealed coke contents of 45.0, 71.2, and 3.2 wt% for the CoFeSi, NiFeSi and FeSi samples, respectively. However, it is necessary to notice the time on stream of each catalyst before the TG analysis.
The catalytic test data, TPD-CO2 measurements, X-ray diffraction and thermal analysis of the spent catalysts were in good agreement and showed that the NiFeSi sample presented higher coke deposition, lower ethylbenzene conversion and higher benzene selectivity. These properties were probably due to the lower CO2 adsorption ability (Fig. 5) and the formation of metallic nickel (Fig. 7) in this sample. Furthermore, these properties could explain the lower selectivity for styrene of this solid.
The thermograms of coked samples exhibited a coke oxidation temperature range of 300 to 450 °C for all the catalysts, although the sample FeSi also showed coke elimination (burning) with an inflection point at 540 °C (Fig. 8), which is related to amorphous carbon.35 Such behaviour (a low temperature burning) was characteristic of either a carbon deposit with a low molecular weight or a low C/H ratio43–45 but also of carbonaceous material containing oxygen in its structure.43,46
To study the molecular structure of the coke deposited on the spent catalysts, the samples were dissolved in concentrated HF and the material (coke) extracted was analyzed by Fourier transformed infrared spectroscopy (FT-IR) (Fig. 9).
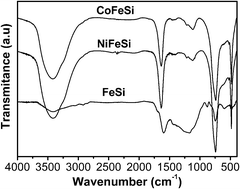 | ||
| Fig. 9 Fourier-transform infrared (FTIR) spectroscopy of material (coke) extracted from the samples, after the catalytic tests. | ||
The spectroscopy results revealed the presence of aliphatic C–H (2918 cm−1 with low intensity) and a band at 1600 cm−1 for frame vibrations of benzene.47 Additionally, a peak at 740 cm−1 was due to the C–H in-plane bending.48 All these peaks illustrated that the precursors of the coke were mainly saturated hydrocarbons.
Further inspecting the FTIR spectrum, a peak at 1400 cm−1 was due to the C–H deformation vibration of a vinyl group bonded to oxygen.49 However, this peak could also be assigned to the CO group of a carboxylate or carbonate-type species.50 The presence of a CO group in the carbonaceous material was reinforced by a peak with low intensity at 1705 cm−1 for FeSi. In addition, the bands at 1200, 1160 and 1110 cm−1 were assigned to OH, COC and CO groups.51 The presence of an OH group in the carbonaceous samples was also suggested by a wide peak at 3418 cm−1; however, the presence of residual water in the carbonaceous sample could not be ruled out.
The coke precursors were mainly composed of aliphatic hydrocarbons as well as oxygen-containing and polyaromatic structures. The adsorption peaks of the FT-IR for aliphatic hydrocarbons and oxygen-containing moieties were much weaker than the peaks for the aromatic hydrocarbons. Therefore, there was a larger amount of aromatic hydrocarbons in the coke precursor. Accordingly, the results from TG and FT-IR analysis suggested that the coke was generated from ethylbenzene but also from CO2.
The observation from the data of TG and FTIR that the coke can be deposited as much of the ethylbenzene as of the carbon dioxide corroborates with the value of ΔG° at 550 °C (−7,9 kJ mol−1) for the reaction (3).
| CO2 + 2H2 ⇌ 2H2O + C | (3) |
In Table 5 are given the relative values of the amount of deposited coke and the ethylbenzene converted for each catalyst, according to the period of reaction time. The data in Table 5 show that the catalyst NiFeSi presents a high ratio of coke formation. Similarly, the data obtained by TG indicate a significant amount of coke in the sample CoFeSi, if compared to the catalyst FeSi.
| Property | Samples | ||
|---|---|---|---|
| CoFeSi | NiFeSi | FeSi | |
| Etbz conv. = total of ethylbenzene converted during the reaction test. X = mass of coke (mg) divided by reaction time and by Etbz conv. (mg). | |||
| Coke/wt% | 45.0 | 71.2 | 3.2 |
| Mass of coke/mg | 122.72 | 370.83 | 4.95 |
| Reaction time/h | 24 | 24 | 6 |
| Etbz conv./mg | 569.86 | 417.53 | 77.27 |
| X | 0.0089 | 0.0370 | 0.0106 |
Conversely, if it is taken into account the reaction time that each catalyst was submitted and the total amount of ethylbenzene converted during this period, it is verified that the ratio of coke deposition for the catalyst CoFeSi is similar (or somewhat lower) to the FeSi sample.
Therefore, although the coke deposition was not suppressed, the addition of Co can be interesting because it can be observed a superior ratio of ethylbenzene conversion without the depreciation of the selectivity for styrene.
Considering the X value from Table 5, the catalytic deactivation (Fig. 6a) could not be attributed mainly to the coke deposition, in spite of the fact that its presence can make difficult the active site accessibility for ethylbenzene.
The catalytic deactivation observed, however, can be attributed mainly to the partial reduction of iron oxide, as suggested by the TPR profile (Fig. 3). As pointed out by Fig. 6a, a higher ratio of catalytic deactivation takes place in the first two hours of time on stream. This behaviour corroborates well with the XRD suggestions, which pointed out that the catalyst samples have a high number of crystalline defects. For the catalytic process a surface with crystalline defects may be more active, because generally a highest surface free energy is observed in surfaces with lower coordination surface atoms. On the other hand, it may present a superior tendency of deactivation.
Accordingly, the XRD profile of the CoFeSi sample after the catalytic test did not pointed out a new phase formation due to the low crystalline diameters or the amorphous characteristic.
4. Conclusions
In this study, cobalt and nickel ferrites dispersed in mesoporous silicon oxide were synthesized by a polymeric precursor method and tested as catalysts for ethylbenzene dehydrogenation in the presence of CO2. The CoFeSi catalyst presented higher ethylbenzene conversion and good styrene selectivity, which were ascribed to the poor reducibility under the reaction conditions and the existence of medium strength basic sites. Conversely, the NiFeSi sample showed higher coke deposition, lower ethylbenzene conversion and high benzene selectivity, probably as a result of the lower CO2 adsorption ability and the formation of metallic nickel during the reaction process for this material. Furthermore, the lower stability observed for the solid FeSi may be correlated with the formation of Fe3O4 during the ethylbenzene reaction. Therefore, in spite of the high coke deposition, the results suggested that materials with a spinel structure, such as ferrites of cobalt, might be particularly useful for ethylbenzene dehydrogenation. On the other hand, as the results suggested that ethylbenzene and CO2 contribute to coke deposition, it is necessary to carry out studies in order to better understand the CO2 role in the ethylbenzene dehydrogenation with CO2.Acknowledgements
The authors acknowledge the “Universidade Federal do Ceará” (UFC), CNPq/CT-PETRO, and Dr J.M. Sasaki (Laboratório de Raios X) for XRD measurements. T.P. Braga, B.M.C. Sales and A.N. Pinheiro express gratitude for scholarships from CNPq and CAPES.References
- E. H. Lee, Catal. Rev.–Sci. Eng., 1973, 8, 285–305 CAS
.
- V. Zhyznevskiy, R. Tsybukh and V. Gumenetskiy, React. Kinet. Catal. Lett., 2000, 71, 209–215 CrossRef CAS
-
K. Othmer, Encyclopedia of Chemical Technology, Willey, New York, 1984, p. 770 Search PubMed
- M. S. Ramos, M. S. Santos, L. P. Gomes, A. Albornoz and M. C. Rangel, Appl. Catal., A, 2008, 341, 12–17 CrossRef CAS
-
A. B. Styles, in Applied Industrial Catalysis, ed. B. E. Leach, Academic Press, New York, 1983, p.137 Search PubMed
- B. D. Herzog and H. F. Rase, Ind. Eng. Chem. Prod. Res. Dev., 1984, 23, 187–196 CrossRef CAS
- N. Mimura and M. Saito, Catal. Today, 2000, 55, 173–178 CrossRef CAS
- G. Carja, Y. Kameshima and K. Okada, Microporous Mesoporous Mater., 2008, 115, 541–547 CrossRef CAS
- T. P. Braga, E. Longhinotti, A. N. Pinheiro and A. Valentini, Appl. Catal., A, 2009, 362, 139–146 CrossRef CAS
- M. Surgino, H. Shimada, T. Turuda, H. Miura, N. Ikenaga and T. Suzuki, Appl. Catal., A, 1995, 121, 125–137 CrossRef
- Y. Ohishi, T. Kawabata, T. Shishido, K. Tanaki, Q. Zhang, Y. Wang and K. Takehira, J. Mol. Catal. A: Chem., 2005, 230, 49–58 CrossRef CAS
- T. P. Braga, A. N. Pinheiro, C. V. Teixeira and A. Valentini, Appl. Catal., A, 2009, 366, 193–200 CrossRef CAS
- R. M. Freire, A. L. Pinheiro, A. C. Oliveira, F. F. de Sousa, A. P. Ayala, J. M. Filho, P. T. C. Freire and A. C. Oliveira, Appl. Catal., A, 2009, 359, 165–179 CrossRef CAS
- A. Miyakoshi, A. Ueno and M. Ichikawa, Appl. Catal., A, 2001, 216, 137–146 CrossRef CAS
- N. Ballarini, F. Cavani, S. Passeri and K. Wilson, Appl. Catal., A, 2009, 366, 184–192 CrossRef CAS
- S. Kameoka, T. Tanabe and A. P. Tsai, Appl. Catal., A, 2010, 375, 163–171 CrossRef CAS
- J. Ak, T. Ghaddar, A. Ghanem and H. El-Rassy, J. Mol. Catal. A: Chem., 2009, 312, 18–22 CrossRef
- E. Manova, T. Tsoncheva, D. Paneva, J. L. Rehspringer, K. Tenchev, I. Mitov and L. Petrov, Appl. Catal., A, 2007, 317, 34–42 CrossRef CAS
- Y. Joseph, M. Wuhn, A. Niklewski, W. Ranke, W. Weiss, C. Woll and R. Schlogla, Phys. Chem. Chem. Phys., 2000, 2, 5314–5319 RSC
- S. M. Montemayor, L. A. García-Cerda, J. R. Torres-Lubián and O. S. Rodríguez-Fernández, Mater. Res. Bull., 2008, 43, 1112–1118 CrossRef CAS
- E. Manova, T. Tsoncheva, Cl. Estournès, D. Paneva, K. Tenchev, I. Mitov and L. Petrov, Appl. Catal., A, 2006, 300, 170–180 CrossRef CAS
- A. A. Mirzaei, A. Beig babaei, M. Galavy and A. Youssef, Fuel Process. Technol., 2010, 91, 335–347 CrossRef CAS
- A. Valentini, N. L. V. Carreño, L. F. D. Probst, E. R. Leite and E. Longo, Microporous Mesoporous Mater., 2004, 68, 151–157 CrossRef CAS
- A. Valentini, N. L. V. Carreño, L. F. D. Probst, A. Barison, A. G. Ferreira, E. R. Leite and E. Longo, Appl. Catal., A, 2006, 310, 174–182 CrossRef CAS
- A. Ries, A. Z. Simões, M. Cilense, M. A. Zaghete and J. A. Varela, Mater. Charact., 2003, 50, 217–221 CrossRef CAS
- J. C. Forti, P. Olivi and A. R. de Andrade, Electrochim. Acta, 2001, 47, 913–920 CrossRef CAS
- C. F. Windisch Jr, K. F. Ferris, G. J. Exarhos and S. K. Sharma, Thin Solid Films, 2002, 420–421, 89–99 CrossRef
- H. M. Rietveld, J. Appl. Crystallogr., 1967, 2, 65–71 CrossRef
- R. A. Young, A. Sakthivel, T. S. Moss and C. O. Paiva-Santos, J. Appl. Crystallogr., 1995, 28, 366–367 CrossRef
- M. I. Mendelson, J. Am. Ceram. Soc., 1969, 52, 443–446 CrossRef CAS
- M. Gotić, G. Koščec and S. Music, J. Mol. Struct., 2009, 924–926, 347–354 Search PubMed
- M. Rajendran, R. C. Pullar, A. K. Bhattacharya, D. Das, S. N. Chintalapudi and C. K. Ajumdar, J. Magn. Magn. Mater., 2001, 232, 71–83 CrossRef CAS
- J. Jacob and M. A. Khadar, J. Appl. Phys., 2010, 107(11), 114310–114310 CrossRef
- J. C. Ryu, D. H. Lee, K. S. Kang, C. S. Park, J. W. Kim and Y. H. Kim, J. Ind. Eng. Chem. (Washington, D. C.), 2008, 14, 252–260 CAS
- B. Pawelec, L. Daza, J. L. G. Fierro and J. A. Anderson, Appl. Catal., A, 1996, 145, 307–322 CrossRef CAS
- J. Guo, Z. Hou, J. Gao and X. Zheng, Fuel, 2008, 87, 1348–1354 CrossRef CAS
- G. A. El-Shobaky, A. M. Turky, N. Y. Mostafa and S. K. Mohamed, J. Alloys Compd., 2010, 493, 415–422 CrossRef CAS
- L. Xiancai, W. Min, L. Zhihua and H. Fei, Appl. Catal., A, 2005, 290, 81–86 CrossRef
- V. R. Choudhary, S. A. R. Mulla and B. S. Uphade, Fuel, 1999, 78, 427–437 CrossRef CAS
- R. Dziembaj, P. K. Strowski and L. Chmielarz, Appl. Catal., A, 2003, 255, 35–43 CrossRef CAS
- C. Cannas, D. Gatteschi, A. Musinu, G. Piccaluga and C. Sangregorio, J. Phys. Chem. B, 1998, 102, 7721–7726 CrossRef CAS
- A. W. Chester, Ind. Eng. Chem. Res., 1987, 26, 863–869 CrossRef CAS
- H. Hattori, Chem. Rev. (Washington, DC, U. S.), 1995, 95, 537–558 CAS
- A. Effendi, K. Hellgardt, Z.-G. Zhang and T. Yoshida, Catal. Commun., 2003, 4, 203–207 CrossRef CAS
- M. Guisnet and P. Magnoux, Appl. Catal., A, 2001, 212, 83–96 CrossRef CAS
- F. Cavani and F. Trifirò, Appl. Catal., A, 1995, 133, 219–239 CrossRef CAS
- Y. He and Y. He, Catal. Today, 2002, 74, 45–51 CrossRef CAS
-
Y. W. Chen, Q. H. Li and W. L. Huang, Organic instrumental analysis, Hunan University Publishing Company, Hunan, 1st edn, 1987 Search PubMed
- S.-H. Wang and P. R. Griffiths, Fuel, 1985, 64, 229236 CrossRef
- W. Wei, J. A. Moulijn and G. Mul, J. Catal., 2009, 262, 1–8 CrossRef CAS
- T. Jiang and K. Xu, Carbon, 1995, 33, 1663–1671 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2011 |