Received
8th April 2011
, Accepted 9th May 2011
First published on 3rd June 2011
Introduction
Selective oxidation is of great importance for the chemical industry especially for the synthesis of chemical intermediates. Selective oxidation is viewed as a valuable pathway for the activation of hydrocarbon feedstocks which can have inherently low activity. Oxygen from the air is considered to be the oxidant of choice and represents the greenest option. Oxygen is a diradical in its ground state which facilitates its involvement in a number of radical reactions which can be useful for a range of low temperature oxidations. However, in many cases more active forms of oxygen have to be used, including non-green stoichiometric oxygen donors such as permanganate and chromates.1 The electrophilic addition of oxygen to an alkene to form an epoxide remains one of the greatest challenges in selective oxidation chemistry. For ethene, the simplest member of this series, catalytic epoxidation has been practiced commercially for decades. Very high selectivities can be achieved using a supported silver catalyst2 but typically chlorinated hydrocarbons and nitrogen oxides have to be added to quench unfavourable by-product formation, and this makes the process non-green in this respect. Higher alkenes cannot be epoxidised with molecular oxygen so readily. Propene can be epoxidised using the titanium silicalite TS-1 as catalyst with hydrogen peroxide.3 Whilst hydrogen peroxide is considered to be a green oxidant, the use of oxygen in the form of air would be preferable in view of the cost differential. In light of these difficulties, there is a quest to find routes for epoxidation of higher alkenes that utilise molecular oxygen.
Recently, Rossi and co-workers4 have reviewed the key aspects concerning selective oxidation. They note the recent advances that have been made using supported gold catalysts. In the mid 1990s Haruta and co-workers were the first to demonstrate the potential of supported gold catalysts for the epoxidation of propene with oxygen in the presence of H2 as a sacrificial reductant that permits the activation of O2 at relatively low temperatures.5,6 In this way it was considered that a hydroperoxy species was formed in situ and this led to the selective oxidation chemistry. Haruta found that Au/TiO2, prepared using deposition precipitation, was selective for propene epoxidation and the catalysis was associated with an intimate contact between hemi-spherical gold nano-crystals (2–5 nm in diameter) and the TiO2 support. Initial selectivities were low but promising; improvements were made by using different titanium-containing supports including TS-1, Ti-zeolite β, Ti-MCM-41 and Ti-MCM-48.7–20 A key issue that remains with this experimental approach is the selectivity based on H2, which can be very low, although recent studies have started to address this problem with some success.21
We have shown that cyclic alkenes can be successfully epoxidised using graphite-supported gold nanoparticles with oxygen as oxidant if catalytic amounts of t-butyl hydroperoxide (TBHP) or H2O2 are added.22 We showed that for the oxidation of cyclooctene using mild, solvent-free conditions, selectivities of over 80% to the epoxide could be achieved. We also showed that the peroxy initiator was not required to achieve oxidation but that lower selectivities to the epoxides were observed in their absence.22 Subsequently, Caps and co-workers23–26 have studied this experimental approach in detail for the oxidation of stilbene, for which they have proposed a radical mechanism. This mechanism is consistent with the initial observation that catalytic amounts of a peroxy species were required to observe selective epoxidation.22 Deng and Friend27 showed using model studies with a Au (111) surface that styrene could be directly epoxidised with oxygen giving 53% styrene oxide. Most recently, Lambert and co-workers28 have shown that very small Au55 nanocrystals supported on carbon are active catalysts for styrene oxidation with oxygen but only minor selectivity to the epoxide was observed and the major product was benzaldehyde. Recently,29 we have explored the reaction conditions required with the graphite-supported gold catalyst to determine how high selectivities can be maintained. It is interesting to note that while heterogeneous gold and gold palladium bimetallic catalysts have been found to be highly effective for oxidation reactions, examples for homogeneous gold catalysts are comparatively rare30 and gold palladium homogeneous catalysts appear to be useful only for cross coupling reactions.31 In this paper we extend our initial studies22,29 to examine the use of graphite-supported gold-palladium alloy catalysts for the selective epoxidation of cyclooctene using oxygen together with catalytic amounts of a peroxy initiator. In addition, we extend these studies to the oxidation of crotyl alcohol using these catalysts, since this substrate presents two oxidisable functional groups, namely a carbon–carbon double bond and a hydroxyl group, and we wished to explore their relative reactivities under mild oxidation conditions.
Experimental
Unless otherwise stated, single metal catalysts (1 wt% Au/support or 1 wt% Pd/support) were prepared using the following standard deposition precipitation method (denoted DP). For the gold catalyst, a solution of HAuCl4·3H2O (5 ml, 2 g in 100 ml distilled water) was diluted with water (45 ml). Aqueous sodium carbonate was added with stirring until pH = 10 was attained. This solution was then added, with continuous stirring, to a slurry of the support in water (4.95 g in water (50 mL)). The mixture was stirred for 1 h at 20 °C, maintaining the pH at 10. The mixture was then heated to 70 °C and formaldehyde was added as a reducing agent unless otherwise stated. The solid was recovered by filtration and washed with water until the washings were found to be chloride free. The catalyst was dried (110 °C, 16 h) prior to use. 1 wt% Pd/support was prepared using the same procedure using a solution of PdCl2 (4.8 mL, 0.5 g in 100 mL distilled water) in place of the gold salt solution. Graphite (Johnson Matthey) and TiO2 (Degussa P25) were used as received as supports. Bimetallic Au–Pd catalysts, comprising 1 wt% total metal, were prepared using the same procedure with appropriate amounts of HAuCl4·3H2O and PdCl2.
Two other preparation methods were also evaluated, namely impregnation and sol-immobilisation. For the impregnation method for a 1 wt% Au catalyst, the support was suspended in distilled water (4.95 g in 100 mL) for 15 min. A solution of HAuCl4·3H2O (5 mL, 2 g in 100 mL distilled water) was added to the slurry slowly dropwise over 30 min. The mixture was stirred under reflux for 30 min; after cooling, formaldehyde was added as a reducing agent. The solid was recovered by filtration and washed with water (1 L) until the washings were found to be chloride free. The catalyst was dried (110 °C, 16 h) prior to use.
For the sol-immobilization method an aqueous solution of HAuCl4·3H2O was prepared. poly(vinyl alcohol) (PVA) (1 wt% solution, Aldrich, MW = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 80% hydrolyzed) was added (PVA/Au (by wt) = 0.65); a freshly prepared solution of NaBH4 (0.1 M, Aldrich, NaBH4/Au (mol/mol) = 5) was then added to form a dark-brown sol. After 30 min, the colloid that had been generated was immobilized by adding the support (acidified at pH 1 by sulfuric acid) with stirring. The amount of support material required was calculated so as to have a total final metal loading of 1 wt%. After 2 h the slurry was filtered, the catalyst washed thoroughly with distilled water (1 L). The catalyst was dried (110 °C, 16 h) prior to use.
000, 80% hydrolyzed) was added (PVA/Au (by wt) = 0.65); a freshly prepared solution of NaBH4 (0.1 M, Aldrich, NaBH4/Au (mol/mol) = 5) was then added to form a dark-brown sol. After 30 min, the colloid that had been generated was immobilized by adding the support (acidified at pH 1 by sulfuric acid) with stirring. The amount of support material required was calculated so as to have a total final metal loading of 1 wt%. After 2 h the slurry was filtered, the catalyst washed thoroughly with distilled water (1 L). The catalyst was dried (110 °C, 16 h) prior to use.
Catalyst testing and characterisation
All reactions were performed in a glass round-bottomed flask (50 ml) fitted with a reflux condenser open to the atmosphere and heated in an oil bath. Typically, cis-cyclooctene (10 mL, 0.077 mol) or crotyl alcohol (10 mL, 0.12 mol) was stirred with a magnetic follower at the desired temperature, then the radical initiator (TBHP, 1.03 × 10−4 mol) was added followed by the catalyst (0.12 g), although it should be noted that the order of addition does not have any effect on the catalytic reaction. The reactions were typically carried out for 24 h. Analysis was carried out using gas chromatography (Varian star 3400 CX) with CP-WAX 52 CB column (25 m length, 0.53 mm O.D. and 0.2 μm film thickness) and a flame ionization detector. Product identification was by GC-MS (Waters, GCT Premier) and 1H NMR spectroscopy (Bruker DPX 400 spectrometer) and by comparison with commercially available samples. Experiments were conducted with at least two separate catalyst preparations and for each catalyst the experiments were carried out in duplicate. The experimental error is ±2% of the quoted values.
X-Ray photoelectron spectra were recorded on a Kratos Axis Ultra DLD spectrometer employing a monochromatic AlKαX-ray source (75–150 W) and analyser pass energies of 160 eV (for survey scans) or 40 eV (for detailed scans). Samples were mounted using double-sided adhesive tape and binding energies referenced to the C(1s) binding energy of adventitious carbon contamination which was taken to be 284.7 eV.
Samples for examination by transmission electron microscopy (TEM) were prepared by dispersing the catalyst powder in high purity ethanol, then allowing a drop of the suspension to evaporate on a holey carbon film supported by a 300 mesh copper TEM grid. Samples were then subjected to bright field diffraction contrast imaging experiments in order to determine particle morphologies and size distributions. The instrument used for this analysis was a JEOL 2000FX TEM operating at 200kV. X-Ray energy dispersive spectroscopy (XEDS) was also carried out in this instrument using an Oxford Instruments INCA XEDS system.
Results and discussion
Effect of catalyst preparation method.
We initially investigated the effect of the preparation method on the oxidation of cis-cyclooctene using graphite-supported catalysts and the results are shown in Table 1. The selectivity we observed was very similar for all preparation methods, namely the main product was the epoxide with ca. 80% selectivity and the major by-product was the allylic alcohol (See Scheme 1).32 Using deposition precipitation or the standard impregnation method did not result in major differences in activity. However, it was noted that there appeared to be no synergistic effect on conversion on addition of Pd to Au for this oxidation reaction. This is in distinct contrast with two other redox reactions we have previously studied in detail, namely the oxidation of primary alcohols33 and the direct synthesis of hydrogen peroxide34 where a very pronounced synergistic effect is observed both on activity and selectivity.
|
Preparation method
|
1%Au |
1% Pd |
1% (Au/Pd) |
| Conv./% |
Sel.% |
Conv.% |
Sel.% |
Conv./% |
Sel.% |
|
Reaction condition: Catalyst (0.12 g), cis-cyclooctene (10 mL, 0.077mol), temperature 80 °C, TBHP (0.01 mL, 1.03 × 10−4 mol), 24 h, atmospheric pressure.
|
|
DP
|
4.6 |
78.9 |
3.1 |
72.3 |
3.3 |
82.9 |
| Immobilization |
7.7 |
81.5 |
2.16 |
79 |
1.6 |
72.7 |
| Impregnation |
4.2 |
71.3 |
2.5 |
75.9 |
4.8 |
76.1 |
In view of this we investigated TiO2-supported catalysts (Table 2) and observed similar results, i.e. no synergistic effect was apparent for the addition of Pd to Au. Again the selectivity to the epoxide was ca. 80% and the allylic alcohol was the major by-product. Consequently, we investigated the effect of the reducing agent, formaldehyde, used in the catalyst preparation on the catalytic activity (Table 3). Improved results were observed when the reducing agent was used compared to its absence.
|
Preparation method
|
1%Au |
1% Pd |
1% (Au/Pd) |
| Conv.% |
Sel.% |
Conv.% |
Sel.% |
Conv.% |
Sel.% |
|
Reaction condition: Catalyst (0.12 g), cis-cyclooctene (10 mL, 0.077mol), temperature 80 °C, TBHP (0.01 mL, 1.03 × 10−4 mol), 24 h, atmospheric pressure.
|
|
DP
|
4.6 |
80.4 |
6.3 |
79.8 |
2.2 |
77.2 |
| Immobilization |
6.3 |
92.8 |
0.27 |
8.4 |
0.15 |
70.4 |
| Impregnation |
1.8 |
74.0 |
0.34 |
85.1 |
3.01 |
77.9 |
|
Catalyst
|
Reduced with formaldehyde |
Unreduced |
Unreduced and calcined at 300 °C |
| Conv.% |
Sel.% |
Conv.% |
Sel.% |
Conv.% |
Sel.% |
|
Reaction conditions: cis-cyclooctene (10 mL, 0.077 mol), catalyst (0.12 g), TBHP (0.01mL 1.03 × 10−4 mol), 80 °C, 24 h, atmospheric pressure.
|
| 1%Au/graphite |
4.0 |
78.2 |
3.7 |
83.0 |
4.6 |
74.9 |
| 1%Pd/graphite |
4.5 |
78 |
3.0 |
70.3 |
5.2 |
71.7 |
| 0.5%Au–0.5%Pd/graphite |
3.6 |
77.7 |
2.6 |
79.6 |
0.84 |
70.1 |
Effect of Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :Pd molar ratio on the epoxidation of cis-cyclooctene
:Pd molar ratio on the epoxidation of cis-cyclooctene
We investigated a range of graphite supported Au–Pd catalysts prepared using the deposition precipitation method all containing 1 wt% of total metal but with varying Au:Pd molar ratios. The effect of this Au:Pd molar ratio on activity is shown in Fig. 1. It is apparent that this variable has a major effect on the observed activity with two minima in activity being observed with Au:Pd molar ratios of 0.74:0.26 and 0.09:0.91 and a distinct maximum at Au::Pd = 0.35:0.65. However, the monometallic Au and Pd catalysts showed marginally higher activity when compared with the bimetallic Au–Pd = 0.35:0.65.catalyst. The selectivity to the epoxide, alcohol, ketone and cyclooctyl hydroperoxide was unaffected by the Au:Pd molar ratio; the epoxide selectivity was ca. 75–80% across the composition range (Fig. 2).
 |
| | Fig. 1 Effect of metal composition on cyclooctene conversion. Reaction conditions: catalyst (0.12 g), cis-cyclooctene (10 mL, 0.077 mol), TBHP (0.01mL, 1.03 × 10−4 mol), temperature 80 °C, glass reactor, 24 h, atmospheric pressure. | |
 |
| | Fig. 2 Effect of metal composition on the selectivity of cyclooctene oxidation. Reaction conditions: catalyst (0.12 g), cis-cyclooctene (10 mL, 0.077 mol), TBHP (0.01mL, 1.03 × 10−4 mol), temperature 80 °C, glass reactor, 24 h, atmospheric pressure. Epoxide (◆), alcohol (■), ketone (▲), hydroperoxide (●). | |
A bright field (BF) micrograph and the particle size distribution for the pure Au/graphite sample are shown in Fig. 3(a) and (b) respectively. The Au particles are isolated and have a median size of 31 nm. Fig. 3(c) and (d) show a typical BF image and the particle size distribution of the pure-Pd/graphite sample. In this case, primary Pd particles with a median size of 6.8 nm are agglomerated into larger chain-like clusters. A sub-set of three Au–Pd/graphite samples were also examined by TEM, namely Au:Pdca. 4:1, 1:1 and 1:4 by weight and the results are shown in Fig. 4, 5 and 6 respectively. The median primary particles sizes for these three Au–Pd samples are 21.7 nm, 16.6 nm and 14.1 nm respectively, and seem to systematically decrease with increasing Pd content. In each case, it is clear that two distinct Au–Pd alloy morphologies exist. The first morphology consists of groups of a few relatively large (20–50 nm) Au–Pd alloy particles (circled in yellow) which were Au-rich. The Au:Pd ratio in these larger particles were rather similar irrespective of the nominal Au:Pd ratio of the sample. The second morphology present was seen to be sizeable agglomerates of much smaller (5–15 nm) Au–Pd alloy particles (circled in red) that are Pd-rich. In this latter case, the Au content was seen to scale systematically with the nominal overall Au content of the sample. No evidence of Au–Pd ordered alloy structures such as those reported by Barabash,35 were found in either of the two morphologies found in any of the Au4Pd1/graphite, Au1Pd1/graphite and Au1Pd4/graphite samples, and this will be studied further in subsequent studies.
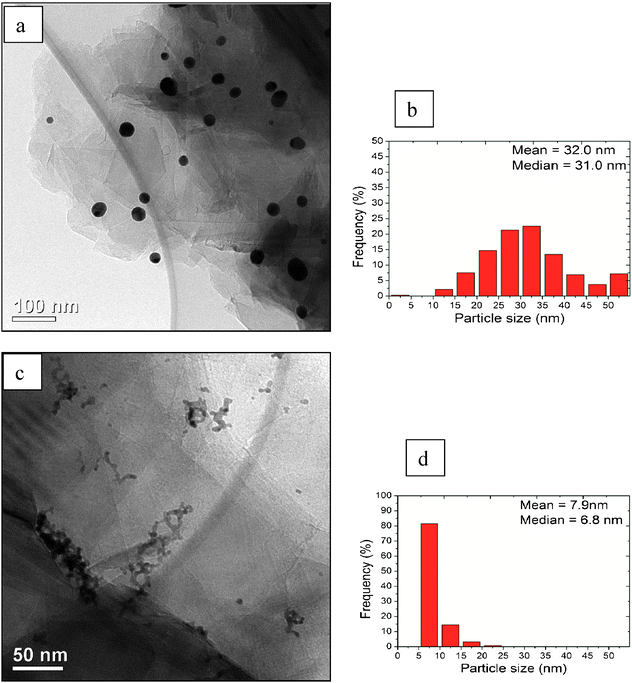 |
| | Fig. 3
TEM and particle size distributions of monometallic catalysts respectively; gold (a) and (b); palladium (c) and (d). | |
 |
| | Fig. 4
TEM, X-EDS and particles size distribution of Au:Pdca. 4:1 by weight (0.74:0.26 molar ratio). | |
 |
| | Fig. 5
TEM, X-EDS and particles size distribution of Au:Pdca. 1:1 by weight (0.35:0.65 molar ratio). | |
 |
| | Fig. 6
TEM, X-EDS and particles size distribution of Au:Pd 1:4 by weight (0.09:0.91 molar ratio). | |
The Au–Pd catalysts were analysed by XPS to determine the mean surface Au:Pd molar ratio and the data are shown in Table 4. It is apparent that this ratio varies with the formal metallic composition and when the activity is at a minimum it coincides with low surface Pd:Au molar ratio (Fig. 7).
Table 4
XPS and theoretical Pd/Au molar ratios for the 1% Au–Pd/C catalysts
|
Au theoretical wt (%) |
Pd Theoretical wt (%) |
Pd theoretical molar (%) |
Theoretical Pd/Au molar ratio |
XPS
Pd/Au corrected molar ratio |
Mean particle size (nm) |
Median particle size (nm) |
Conversion (%) |
| 1 |
0 |
0 |
0 |
0 |
32.0 |
31.0 |
4.0 |
| 0.9875 |
0.0125 |
0.02 |
0.02 |
0.02 |
— |
— |
3.8 |
| 0.837 |
0.163 |
0.26 |
0.36 |
0.91 |
23.1 |
21.7 |
1.5 |
| 0.5 |
0.5 |
0.65 |
1.86 |
6.38 |
17.9 |
16.6 |
3.6 |
| 0.163 |
0.837 |
0.91 |
9.54 |
6.22 |
16.0 |
14.5 |
2.2 |
| 0.0125 |
0.9875 |
0.99 |
146.2 |
41.1 |
— |
— |
4.2 |
| 0 |
1 |
1 |
∞ |
— |
7.9 |
6.8 |
4.5 |
 |
| | Fig. 7 The influence on the cyclooctene conversion of the mean molar% of surface Pd determined by XPS Reaction conditions: catalyst (0.12 g), cis-cyclooctene (10 mL, 0.077 mol), TBHP (0.01mL, 1.03 × 10−4 mol), temperature 80 °C, glass reactor, 24 h, atmospheric pressure. | |
We have studied the reaction over time (Fig. 8) to investigate whether the epoxide selectivity changes with increasing conversion for the different Au:Pd ratios. Initially the epoxide selectivity is lower with AuPd catalysts when compared to the monometallic Au catalyst. With the Pd containing catalysts the selectivity to the allylic alcohol is high at short reaction times. However, after 24 h reaction the epoxide selectivity improves and is comparable at ca. 80% (Fig. 9). We consider that the pathway outlined by Bawaked et al.32 initially proceeds through the cyclooctenyloxy radical which is able to abstract hydrogen from the allylic position of cyclooctene to form the alcohol or attack the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of a cyclooctene molecule to produce an adduct that fragments to form the epoxide. The former route is apparently restricted as the reaction proceeds as observed with the monometallic Au catalyst.
C bond of a cyclooctene molecule to produce an adduct that fragments to form the epoxide. The former route is apparently restricted as the reaction proceeds as observed with the monometallic Au catalyst.
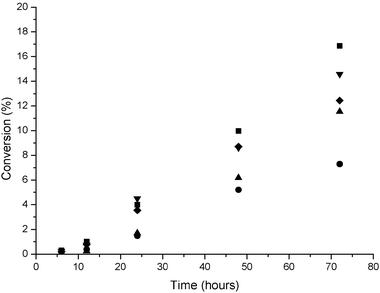 |
| | Fig. 8 Effect of reaction time on cis-cyclooctene conversion. 1 wt% Au/graphite (■), 0.84 wt% Au–0.16 wt% Pd/graphite (●), 0.72 wt% Au–0.28 wt% Pd/graphite (▲), 0.5 wt% Au 0.5 wt% Pd/graphite (◆), 1 wt% Pd/graphite (▼). Reaction conditions: catalyst (0.12 g), cis-cyclooctene (10 mL, 0.077 mol), TBHP (0.01mL, 1.03 × 10−4 mol), temperature 80 °C, atmospheric pressure. | |
 |
| | Fig. 9 Effect on selectivity over time to epoxide (= closed symbol) and alcohol (= open symbol) from cis-cyclooctene oxidation with different catalysts; .1 wt% Au/graphite (■), 0.84 wt% Au–0.16 wt% Pd/graphite (●), 0.72 wt% Au–0.28 wt% Pd/graphite (▲), 0.5 wt% Au 0.5 wt% Pd/graphite (◆), 1 wt% Pd/graphite (▼). Reaction conditions: catalyst (0.12 g), cis-cyclooctene (10 mL, 0.077 mol), TBHP (0.01mL, 1.03 × 10−4 mol), temperature 80 °C, atmospheric pressure. | |
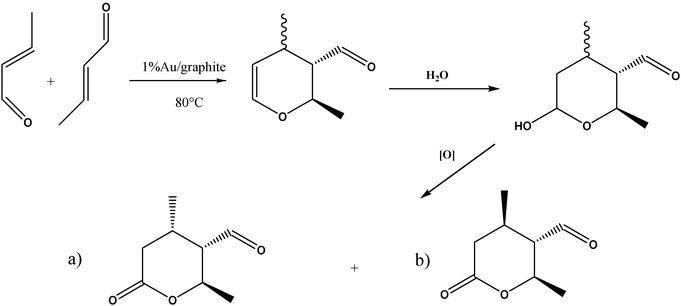
Crotyl alcohol oxidation was investigated using 1 wt% Au supported on graphite prepared using the deposition precipitation and impregnation methods and the results are shown in Table 5. We selected crotyl alcohol as there are two functional groups present that can be oxidised (Scheme 1). Previous studies by Della Pina et al.36 have shown that allyl alcohol can be selectively converted to 3 hydroxy propionic acid with a Au/C catalyst, hence we wanted to investigate if the carbon–carbon double bond could be epoxidised using our reaction conditions. In these initial experiments we used the same reaction conditions that we had adopted for the epoxidation of cis-cyclooctene, and in particular TBHP was present as a radical initiator in catalytic amounts. Both methods gave very similar activities and it should be noted that when tested under similar conditions this substrate was more reactive (2 to 17% conversion after 24 h) than cis-cyclooctene (2 to 4% conversion after 24 h) when Pd is present (supplementary information Fig. S1 and Fig. 1 respectively). The major product from these reactions of crotyl alcohol 1, crotonaldehyde 2, is most obviously a primary oxidation product (Scheme 2, 3). However, this and other observed products could also arise by π-allyl formation when palladium is present. Hydration of such a species adjacent to the hydroxyl group would also generate crotonaldehyde 2, while addition at the opposite end would lead to 3-hydroxybutanal, which was not isolated. However, this could subsequently undergo dehydration to give crotonaldehyde 2 or retro-aldol fragmentation to give two molecules of acetaldehyde, which also was not observed. However, rapid oxidation of this aldehyde would be expected which would then explain the formation of acetic acid 9 (Scheme 4). The implication that a π-allyl species is involved would also account for the formation of both 3-buten-1-ol 3, another significant product, and of butanal 5, following alkene isomerisation without hydration. Subsequent oxidation of the former would then lead to the relatively small amount of butanoic acid 6 observed, as would formation of crotonic acid 10, by direct oxidation of crotonaldehyde 2.
 |
| | Scheme 2 Principal pathways for transformation of trans-crotyl alcohol in air catalysed by Au and Au–Pd on graphite. | |
 |
| | Scheme 3 Reaction network of crotyl alcohol (1) transformation to oxidation and isomerisation products under mild oxidising conditions. | |
A possible origin of the keto-alcohol 4 is by a Wacker-type oxidation37–39 of 3-buten-1-ol 3, but could conceivably also arise by rearrangement of the epoxide derived from crotyl alcohol. However, no epoxide or other obvious products arising from it were observed; indeed, we conclude that epoxidation of the starting alcohol 1 appears not to occur to any detectable level. An alternative pathway to the keto-alcohol 4 may also involve π-allyl hydration, along the lines outlined above. The two diols, 7 and 8, probably arise from metal-catalysed alkene hydration of the initial crotyl alcohol 1. Finally, a pair of initially unknown compounds, which had similar GC retention times, was tentatively assigned as the epimeric valerolactones 11 (Scheme 3 and 5 (a) lactone 1 and (b) lactone 2), which could be formed by a formal Diels–Alder dimerisation of crotonaldehyde 2, a known reaction40 which occurs under acid- or base-catalysis (Scheme 5). In the present case, Lewis-acid catalysis could operate although a plausible alternative is a radical (or base-)-induced Michael addition-trap mechanism, initiated by the conjugate addition of a radical or nucleophilic species to crotonaldehyde. Mass spectrometric evidence suggests that the initial dihydropyran undergoes hydration and oxidation to arrive at the lactones 11 and hence this assignment must be regarded as tentative as it is based solely on such MS evidence.
Some support for these series of mechanisms was obtained by subsequent experiments (Table S1). Firstly, exposure of acetaldehyde to typical reaction conditions gave predominant conversion to acetic acid 9 (85% in 12 h at 74% conversion) (as shown in Scheme 4). Similarly, exposure of pure crotonaldehyde to similar conditions gave, at around 10% conversion, a mixture of the two epimers 11 (∼30%), together with 21% acetic acid 9. The latter could have arisen from Michael addition of water and subsequent retro-aldol fragmentation of the resulting 3-hydroxybutanal and oxidation of the acetaldehyde so formed. A low conversion (ca. 1%) of 3-buten-1-ol 3 into the keto-alcohol 4 was also observed, so the proposed relationship between these two products remains somewhat tentative.
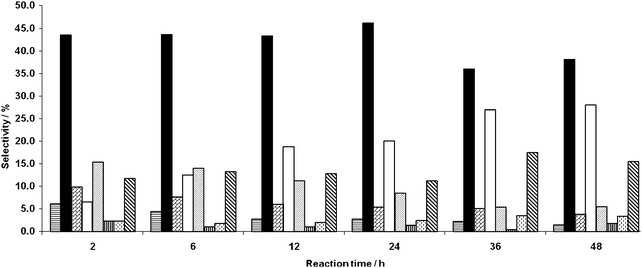
We then investigated the effect of the Au:Pd molar ratio on the activity and selectivity for the reaction of crotyl alcohol. We used catalysts prepared by the deposition precipitation method all containing 1 wt% of total metal but with varying Au:Pd molar ratios and the effect of the Au:Pd molar ratio on product distribution at 24 h is shown in Fig. 10. The product yield increases steadily to a maximum at 0.65 molar ratio as Pd is added to the catalyst composition. The observation of this clear synergistic effect with an optimal Au–Pd ratio occurs with this substrate unlike with cyclooctene. The origin of this type of synergistic effect has been discussed previously33,34 and we consider it to be due to an electronic effect due to alloy formation. The effect on the selectivity was very marked (Fig. S2). Note that the fastest rates (highest conversions) are associated with high selectivity to but-3-en-1-ol, suggesting that the high catalyst activity is associated with isomerisation, which swamps a relatively invariant oxidation rate for the alcohol function (Fig. 10). Presumably these two processes can outrun crotyl alcohol epoxidation, which is likely to be slower than epoxidation of cis-cyclooctene. We cannot, however, rule out epoxidation totally, since we have not checked the stability of the epoxide under the reaction conditions and some of the products (e.g.4-hydroxy-2-butanone) could be derived from the epoxide by known rearrangement processes. With catalyst comprising mainly Pd, the isomerisation reaction was dominant and modest levels of oxidation were observed. Additionally, 4-hydroxy-2-butanone was detected in significant quantities when Pd was present. In the absence of independent evidence for epoxide formation, it is possible that it arises via a Wacker-like oxidation37–39 of 3-buten-1-ol, since the 4-hydroxy-2-butanone selectivity increases monotonically throughout the reaction period, while the 3-buten-1-ol selectivity passes through a maximum after a few hours and then declines, as expected for a sequential isomerisation and oxidation. Oxidation of the alcohol group was observed with all catalyst formulations, however, where gold is higher in surface concentration the yield of crotonaldehyde is increased (Fig. 10). The exception to this is the Pd only catalyst which has a higher selectivity to crotonaldehyde than the Au–Pd catalysts. Gold, we consider, promotes/favours alcohol oxidationvia O2 or a radical species derived from it on the metal surface. Similarly, with the pure Pd catalyst, the incidence of oxidation increased although isomerisation remained dominant. This indicates that the Pd and Au–Pd catalysts facilitate isomerisation, which we suggest may be due to the generation of π-allyl intermediates by chemisorption with allylic C–H dissociation at C-1 and C-3. The production of acetic acid is apparently unfavourable when even minor quantities of Pd are present.
 |
| | Fig. 10 Effect of metal composition on the product yield of crotyl alcohol conversion. Reaction conditions: Catalyst (0.12 g), crotyl alcohol (10 mL, 0.12 mol), temperature 80 °C, TBHP (0.01 mL, 1.03 × 10−4 mol), 24 h, atmospheric pressure. Butyraldehyde ( ), crotonaldehyde ( ), crotonaldehyde ( ), 3-buten-1-ol ( ), 3-buten-1-ol ( ), acetic acid ( ), acetic acid ( ), lactone epimer 1 ( ), lactone epimer 1 ( ), 4-hydroxy-2-butanone ( ), 4-hydroxy-2-butanone ( ), lactone epimer 2 ( ), lactone epimer 2 ( ), butyric acid ( ), butyric acid ( ). ). | |
With respect to the differences observed after 24 h of reaction, we extended the study to encompass a reaction profile over 48 h for an Au and an Au–Pd catalyst (Fig. 11). The rate of crotyl alcohol conversion is ca. 7 times greater with the Au–Pd catalyst (0.153 mmol h−1) compared to the Au catalyst (0.021 mmol h−1). The selectivity to the oxidation product was found to be high with the Au catalyst, however, acetic acid concentration increased over this time period (Fig. 12). Compared to the Au–Pd catalyst, the selectivity was observed to be high with respect to the isomerisation product initially (Fig. 13). However, the formation of two valerolactone epimers (Scheme 5) as secondary products was found to increase over this time period. The implication of these observations can be summarised thus; over Au, crotyl alcohol is first oxidised to crotonaldehyde and/or subsequently transformed into acetic acid. As a minor pathway the catalyst facilitates conversion of the oxidation product crotonaldehyde to the postulated epimers. Likewise over the Au–Pd catalyst the pathway includes firstly isomerisation to 3-buten-1-ol and thereafter, Wacker-like oxidation to 4-hydroxy-2-butanone.
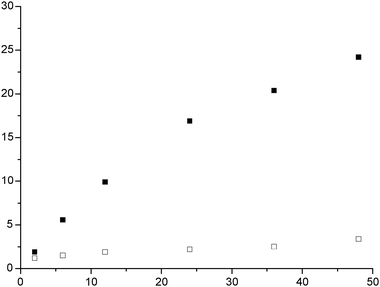 |
| | Fig. 11 Effect of reaction time on the conversion of crotyl alcohol using 0.5 wt%Au–0.5 wt%Pd/graphite (■) or 1 wt%Au/graphite (□) as catalysts. Reaction conditions: Catalyst (0.12 g), crotyl alcohol (10 mL, 0.12mol), temperature 80 °C, TBHP (0.01 mL, 1.03 × 10−4 mole), atmospheric pressure. | |
 |
| | Fig. 12 Selectivity to products of crotyl alcohol conversion using 1 wt%Au/graphite as catalyst. Reaction conditions: Catalyst (0.12 g), crotyl alcohol (10 mL, 0.12 mol), temperature 80 °C, TBHP (0.01 mL, 1.03 × 10−4 mol), atmospheric pressure. Butyraldehyde ( ), crotonaldehyde ( ), crotonaldehyde ( ), 3-buten-1-ol ( ), 3-buten-1-ol ( ), acetic acid ( ), acetic acid ( ), lactone epimer 1 ( ), lactone epimer 1 ( ), 4-hydroxy-2-butanone ( ), 4-hydroxy-2-butanone ( ), butyric acid ( ), butyric acid ( ), lactone epimer 2 ( ), lactone epimer 2 ( ). ). | |
 |
| | Fig. 13 Selectivity to products of crotyl alcohol conversion using 0.5 wt%Au–0.5 wt%Pd/graphite as catalyst. Reaction conditions: Catalyst (0.12 g), crotyl alcohol (10 mL, 0.12 mol), temperature 80 °C, TBHP (0.01 mL, 1.03 × 10−4 mol), atmospheric pressure. Butyraldehyde ( ), crotonaldehyde ( ), crotonaldehyde ( ), 3-buten-1-ol ( ), 3-buten-1-ol ( ), acetic acid ( ), acetic acid ( ), lactone epimer 1 ( ), lactone epimer 1 ( ), 4-hydroxy-2-butanone ( ), 4-hydroxy-2-butanone ( ), butyric acid ( ), butyric acid ( ), lactone epimer 2 ( ), lactone epimer 2 ( ). ). | |
As we observed no significant formation of the epoxide, even at higher TBHP loadings (0.5 mL, 5.16 × 10−4 moles), we investigated the reaction of crotyl alcohol in the absence of the catalytic amount of TBHP and we observed that this made no difference to the observed catalysis (Fig. S3, S4). This is consistent with our previous studies where we have shown that primary alcohols can be oxidised to aldehydes using molecular oxygen in the absence of radical initiators.30 Hence, the presence of the alcohol functional group essentially stops the possibility of the epoxidation pathway under our conditions. Furthermore, previous studies on the oxidation of crotyl alcohol with titania-silicate materials and H2O2 with mild conditions41 detail subsequent oxidation products which we do not detect under our conditions. These secondary products are triol and ether diols, which typically build up when reaction times are extended over 24 h.
Conclusions
We have extended the study of cis-cyclooctene epoxidation using mild oxidising conditions to investigate catalyst preparative methods. The results indicate that although no synergistic effects between Au and Pd were noted with cis-cyclooctene, modest improvements in epoxide yield could be achieved through careful catalyst preparation. Investigating these AuPd mixed catalysts for cis-cyclooctene oxidation has revealed the complex nature of such mixtures, through TEM and XPS, and their influence on the activity observed. The interesting activity profile generated under these conditions may suggest that structured alloys were produced, as the minima correspond to regions where the ratio of Au:Pd is 1:4 and 4:1 by weight, but at present detailed evidence for their formation has yet to be obtained. Examination by electron microscopy and XPS indicate that the particle size and the mean surface Pd concentration combine to affect the activity observed so that, where the surface Pd concentration is highest, the activity is correspondingly high.
Oxidation of crotyl alcohol under similar conditions resulted in a larger range of products wherein the influence of Au and Pd could be clearly differentiated. Where Au was present in high concentrations, oxidation to crotonaldehyde was the preferred pathway. However, when Pd was present in the catalyst the dominant pathway was isomerisation followed, we suggest, by Wacker-like oxidation. The conversion of crotyl alcohol is highly dependent on the ratio of Au and Pd. Monometallic Au was observed to have a low activity, whereas mixtures of Au and Pd had the highest activity. This synergistic effect has been observed before with other catalysts and appears to be related, at least in part to the isomerisation of the starting material when Pd is present. The mechanistic pathway for both catalysts studied is represented in the reaction networks we have provided.
Acknowledgements
This work formed part of an EPSRC funded project and we thank them for funding this research. We also thank King Abdul Aziz University (Saudi Arabia Government) for financial support. We thank R. Jenkins for assistance with product identification.
References
- R. A. Sheldon, Stud. Surf. Sci. Catal., 1991, 66, 33.
- R. M. Lambert, F. J. Williams, R. L. Cropley and A. Palermo, J. Mol. Catal. A: Chem., 2005, 228, 27 CrossRef CAS.
- E. Klemm, E. Dietzsch, T. Schwarz, T. Kruppa, A. Lange de Oliveira, F. Becker, G. Markowz, S. Schirrmeister, R. Schütte, K. J. Caspary, F. Schüth and D. Hönicke, Ind. Eng. Chem. Res., 2008, 47, 2086 CrossRef CAS.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077 RSC.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566 CrossRef CAS.
- M. Haruta and M. Date, Appl. Catal., A, 2001, 222, 427 CrossRef CAS.
- T. A. Nijhuis, T. Visser and B. M. Weckhuysen, J. Phys. Chem. B, 2005, 109, 19309 CrossRef CAS.
- B. S. Uphade, S. Tsubota, T. Hayashi and M. Haruta, Chem. Lett., 1998, 1277 CrossRef CAS.
- T. A. Nijhuis, B. J. Huizinga, M. Makkee and J. A. Moulijn, Ind. Eng. Chem. Res., 1999, 38, 884 CrossRef CAS.
- T. A. Nijhuis, T. Q. Gardner and B. M. Weckhuysen, J. Catal., 2005, 236, 153 CrossRef CAS.
- T. A. Nijhuis, T. Visser and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2005, 44, 1115 CrossRef CAS.
- T. A. Nijhuis and B. M. Weckhuysen, Chem. Commun., 2005, 6002 RSC.
- N. Yap, R. P. Andres and W. N. Delgass, J. Catal., 2004, 226, 156 CrossRef CAS.
- A. Zwijnenburg, M. Makkee and J. A. Moulijn, Appl. Catal., A, 2004, 270, 49 CrossRef CAS.
- C. Qi, T. Akita, M. Okumura, K. Kuraoka and M. Haruta, Appl. Catal., A, 2003, 253, 75 CrossRef CAS.
- B. Taylor, J. Lauterbach and W. N. Delgass, Appl. Catal., A, 2005, 291, 188 CrossRef CAS.
- A. K. Sinha, S. Seelan, M. Okumura, T. Akita, S. Tsubota and M. Haruta, J. Phys. Chem. B, 2005, 109, 3956 CrossRef CAS.
- B. Chowdhury, J. J. Bravo-Suarez, M. Date, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2006, 45, 412 CrossRef.
- E. Sacaliuc, A. M. Beale, B. M. Weckhuysen and T. A. Nijhuis, J. Catal., 2007, 248, 235 CrossRef CAS.
- J. Lu, X. Zhang, J. J. Bravo-Suárez, K. K. Bando, T. Fujitani and S. T. Oyama, J. Catal., 2007, 250, 350 CrossRef CAS.
- A. K. Sinha, S. Seelan, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 1546 CrossRef CAS.
- M. D. Hughes, Y.-J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132 CrossRef CAS.
- P. Lignier, F. Morfin, S. Mangematin, L. Massin, J.-L. Rousset and V. Caps, Chem. Commun., 2007, 186 RSC.
- P. Lignier, S. Mangematin, F. Morfin, J.-L. Rousset and V. Caps, Catal. Today, 2008, 138, 50 CrossRef CAS.
- P. Lignier, F. Morfin, Laurent Piccolo, J.-L. Rousset and V. Caps, Catal. Today, 2007, 122, 284 CrossRef CAS.
- D. Gajan, K. Guillois, P. Delichère, J.-M. Basset, J.-P. Candy, V. Caps, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2009, 131, 14667 CrossRef CAS.
- X. Deng and C. M. Friend, J. Am. Chem. Soc., 2005, 127, 17178 CrossRef CAS.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981 CrossRef CAS.
- S. Bawaked, N. F. Dummer, N. Dimitratos, D. Bethell, Q. He, C. J. Kiely and G. J. Hutchings, Green Chem., 2009, 11, 1037 RSC.
- A. S. K. Hashmi, C. Lothschuetz, M. Ackermann, R. Doepp, S. Anantharaman, B. Marchetti, H. Bertagnolli and F. Rominger, Chem.–Eur. J., 2010, 16, 8012 CAS.
- A. S. K. Hashmi, C. Lothschutz, R. Dopp, M. Rudolph, T. D. Ramamurthi and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8243 CrossRef CAS.
- S. Bawaked, N. F. Dummer, D. Bethell, D. W. Knight and G. J. Hutchings, Green Chem., 2011, 13, 127 RSC.
- D. Enache, J. K. Edwards, P. Landon, B. Solsona, A. Carley, A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS.
- J. K. Edwards, B. Solsona, E. Ntainjua, A. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037 CrossRef CAS.
- S. V. Barabash, V. Blum, S. Müller and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 035108 CrossRef.
- C. Della Pina, E. Falleta and M. Rossi, ChemSusChem, 2009, 2, 57 CrossRef.
- J. Tsuji, H. Nagashima and H. Nemoto, Org. Synth., 1984, 62, 9 CAS.
- H. Pellissier, P.-Y. Michellys and M. Santelli, Tetrahedron Lett., 1994, 35, 6481 CrossRef CAS.
- T. Yokota, A. Sakakura, M. Tani, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 2002, 43, 8887 CrossRef CAS.
-
(a) A. Losse, Chem. Ber., 1967, 100, 1266 Search PubMed;
(b) D. W. Cameron and P. E. Schuetz, J. Chem. Soc. (C), 1968, 1801–1802 RSC.
- L. J. Davies, P. McMorn, D. Bethell, P. C. Bulman Page, F. King, F. E. Hancock and G. J. Hutchings, J. Mol. Catal. A: Chem., 2001, 165, 243 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.