High yield synthesis of novel boron nitride submicro-boxes and their photocatalytic application under visible light irradiation
Meng
Wang
a,
Menghua
Li
b,
Liqiang
Xu
*a,
Liancheng
Wang
a,
Zhicheng
Ju
a,
Guangda
Li
a and
Yiti
Qian
a
aKey Laboratory of Colloid and Interface Chemistry, Shandong University, Ministry of Education, Jinan 250100, PR China. E-mail: xulq@sdu.edu.cn; Fax: +86-531-8836-6280; Tel: +86-531-8836-6280
bDepartment of Chemistry and Chemical Engineering, Qilu Normal University, Jinan 250013, PR China
First published on 11th July 2011
Abstract
In this study, hexagonal boron nitride submicro-boxes (BNMB) (0.50–1.4 μm) have been synthesized by using KBH4, NH4F and Zn in a stainless steel autoclave at 450 °C. The formation process was studied by XRD, TEM and EDS, and it is considered that the in situ formed KZnF3 intermediate cubes serve as templates for the formation of BNMB. The as-formed BNMB, with unique structural features, high specific surface area (∼86.9 m2 g−1) and good chemical properties, can be applied as a catalyst support for SnO2. The UV-Vis diffuse reflectance spectrum of SnO2/BNMB shows the absorption edge in the visible region (∼470 nm), making it suitable for photocatalytic application. The experimental result indicates that the SnO2/BNMB exhibited excellent photocatalytic activity on the degradation of methyl orange (MO), which was up to 92% after 30 min of visible-light (λ > 420 nm) irradiation. The good photocatalytic activity was attributed mainly to its suitable band gap energy, strong adsorption ability for MO, and effective charge separation at the SnO2/BNMB photocatalyst interface.
1 Introduction
Owing to their unique properties and wide promising applications in encapsulation and controlled release of materials, robust catalysts and carriers, chemical and biological sensing, and energy-storage media, many efforts have been dedicated to the fabrication of micro- and nanobox materials.1–3 Therefore, various hollow box materials such as Au, Pt, Cu, CuxS, SnO2, Co3O4, Fe(OH)x, WS2, C, PbSe, HZSM-5 and CaTiO3 have been successfully prepared.4–15 Hexagonal boron nitride (h-BN) boxes may be an ideal candidate for functional materials due to their distinct advantages in comparison to other materials. As an important III–V compound, h-BN exhibits a series of remarkable properties like superb thermal stability and oxidation resistance, good mechanical strength and chemical inertness, unique luminescence and biocompatibility,16–18 which can be quite important for a wide range of technical applications. Furthermore, for its high void ratio, large specific surface area and light weight, h-BN with hollow interiors has many potential/demonstrated applications, such as protective shields for materials, carriers for functional species, in building skeletons to construct devices, biological probes or catalyst supports. Accordingly, considerable effort has been directed toward the synthesis and functionalization of various BN hollow structures, e.g. micro-/nanotubes, nanoribbons, nanocages and hollow spheres.19–22 However, for h-BN box structures, especially BN microboxes with well-defined hollow interiors, large void and high specific surface areas have not yet been well studied, which still remains a challenge to material researchers.Tin oxide (SnO2) is a promising photocatalyst on account of its excellent photoelectronic properties and physicochemical stabilities. However, pure SnO2 only exhibits photocatalytic activities under UV irradiation due to its large band gap (Eg = 3.6 eV). Recently, nanoporous SnO2 and SnO2 bipods have been prepared and they have shown visible light photocatalysis for organic dye due to their distinctive particle form.23,24 A drawback of these materials is that the recovery of nanosized photocatalysts in the aqueous phase has been greatly limited, for the separation of fine particles from treated water is usually difficult. To overcome this problem and improve photocatalytic performance of SnO2, current studies hereby should focus on the immobilization of the SnO2 photocatalyst onto appropriate supports.
Recently, catalytic active phases loaded on promising supports could create opportunities for substantial progress in numerous fields, and h-BN is one of these significant supports for catalysts. There are several reports published about the good performance of boron-nitride-based catalysts for catalytic applications, including deep oxidation of volatile organic compounds, hydrogen production, ammonia synthesis, NOx reduction or selective hydrogenation, etc.21,25–28 because of their low density, excellent chemical inertness and environmental friendliness. In addition, particular advantages of h-BN are its layered structure and hydrophobic nature. Several reports demonstrate that photocatalysts loaded on the high surface area supports with layered structure or hydrophobic property, such as SnO2/MgAl-layered double hydroxide, TiO2/Si3N4, and TiO2/hydrophobic mesoporous Si,29–31 show enhanced photocatalytic activity for degrading organic pollutants in an aqueous system. That may be because the hydrophobic support possesses favourable absorption ability for organic contaminants. Likewise, layered or high surface area materials exhibit the high adsorption capacity for organic dyes, available for enhanced photocatalytic reactivity. Moreover, a photocatalyst loaded on a layered structure is beneficial for reducing the rapid recombination of photogenerated electron–hole pairs which are necessary for the photocatalytic reaction.29,32 However, the above reports sometimes involve complicated experimental processes, toxic or dangerous starting materials, limited chemical stability, and the related photocatalyst only active under UV irradiation. Thus, having in mind all the mentioned advantages of BNMB, it is of great interest to consider that BNMB could be advantageously used as a photocatalyst support for SnO2.
In this study, the fabrication of a novel BNMB with a high yield was fulfilled by using KBH4, NH4F and Zn in a stainless steel autoclave at 450 °C. The edge length of the BNMB was in the range of 0.50–1.4 μm and the thickness was of about 20–30 nm. According to the fact that solid cubes (TEM observation), KZnF3 phase (XRD pattern) as well as K, Zn, F elements (EDS spectrum) were detected in the crude samples, an in situ template role of cubic KZnF3 was considered. The total surface area of the BNMB calculated from the analysis of N2 adsorption isotherm data is ∼86.9 m2 g−1, suitable for using as a catalyst support. Therefore, in the second part of the work, the SnO2 photocatalyst was deposited on BNMB by a simple wet chemistry method. The as-formed SnO2/BNMB has the absorption edge at ∼470 nm corresponding to the band gap energy of 2.4 eV, making it suitable as an effective photocatalyst for photocatalytic application. And the photocatalytic performance of SnO2/BNMB has been investigated via photocatalytic degradation of MO under visible irradiation.
2 Experimental
2.1 Synthesis
All the chemical reagents were of analytical-grade purity (Shanghai Chem. Co.) and were used without further purification. In a typical synthesis process, KBH4 (0.015 mol), NH4F (0.105 mol) and Zn powder (0.045 mol) were mixed and put into a 20 mL stainless steel autoclave. The autoclave was sealed and heated from room temperature to 450 °C with a heating rate of 10 °C min−1, and then it was maintained at the target temperature for 30 h. After the autoclave was cooled to room temperature naturally, the crude product was collected and washed with dilute hydrochloric acid and distilled water several times to remove impurities. Finally, the product was dried in a vacuum at 80 °C for 6 h. To date, there has been no research on the preparation and catalytic properties of the SnO2 photocatalyst loaded on BNMB. Here, a common wet chemistry method was applied to the preparation of SnO2/BNMB composites.33SnCl2 powders (1.6 g, anhydrous, Alfa, 99%) were dissolved in distilled water (100 mL), and then HCl (1.2 mL, 38%) and BNMB (0.3 g) were added, respectively. The mixed solution was stirred and refluxed at 100 °C, and maintained at this temperature for 5 h. As the system was cooled to room temperature naturally, the as-formed sample was collected and washed with distilled water to a neutral pH. The as-formed SnO2/BNMB composites were obtained after they were dried in a vacuum at 80 °C for 6 h and finally collected.2.2 Characterization
The products were determined by a Bruker D8 advanced X-ray diffractometer with Cu-Kα radiation, Fourier transformation infrared spectroscopy (FTIR, VERTEX-70), and the Raman spectrum (NEXUS 670). To observe the morphology and structure of the samples, high-resolution electron microscopy (HRTEM, JEOL-2100) and field emission scanning electron microscopy (FESEM, JEOL JSM-7600F) were performed. Thermal gravimetric analysis (TGA) was performed on a Mettler Toledo TGA/SDTA 851 thermal analyzer apparatus under an ambient atmosphere. The BET surface area (SBET) and Barrett–Joyner–Halenda (BJH) pore size distribution (PSD) were measured using a QuadraSorb SI surface area analyzer. The optical absorption spectrum was conducted by a UV-Vis-NIR spectrometer (Shimadzu, UV-1700), and the CL spectrum was collected at room temperature with a Hitachi S4200 instrument. A diffuse reflectance UV-Vis spectrophotometer (DRS, TU-1901) was used to obtain the optical absorption spectra of photocatalysts and mass spectrometry (Agilent 6510 Q-TOF MS) was used to monitor the degradation process of MO.2.3 Catalytic measurement
The photocatalysis experiments of the SnO2/BNMB were carried out in a 100 mL cylindrical Pyrex glass reactor which contains 50 mL (25 mg L−1) of MO aqueous solution and 0.025 g of photocatalysts. First, the suspension was stirred under dark conditions for 30 min to establish an adsorption–desorption equilibrium. Then, the aqueous suspension was irradiated under a 300 W xenon lamp (PLS-SXE300) which has an UV-cut-off filter as the visible light source (irradiation in the range of λ > 420 nm). Analytical samples were taken from the reaction systems every ten minutes and centrifuged to separate photocatalysts before analysis. The characteristic absorption of MO at λ = 464 nm is selected to monitor the photocatalytic degradation process, and the rate of degradation is estimated from residual concentration spectrophotometrically.3 Results and discussion
The typical XRD pattern of the sample obtained at 450 °C for 30 h is shown in Fig. 1a. All the diffraction peaks can be indexed as hexagonal boron nitride, the calculated lattice constant (a = 2.501 Å, c = 6.669 Å) is in accord with the reported value (JCPDS card no. 34-0421). It is worth noting that the Bragg diffraction peak (002) shifts slightly to a lower angle compared to that of the well-crystalline h-BN, which indicates lattice expansion between two adjacent BN slabs along the c-axis. That could be attributed to the introduction of the strain owing to the curvature of the h-BN layers.34 Meanwhile, the broadening of the (002) peak is caused by the coherent X-ray scattering of the reduced domain size in the direction that is perpendicular to the (00l) plane. Fig. 1b shows the typical FTIR spectrum of the sample. Two main characteristic absorption bands can be observed around 1377 and 814 cm−1. The former can be attributed to the stretching vibration of the B–N bond, while the latter belongs to the B–N–B out-of-plane bending vibrations.35 The weak absorption bands near 3420 and 3400 cm−1 result from the residual N–H stretching and O–H vibrations that adsorbed on the sample surface. The corresponding Raman spectrum of the sample displays a dominant peak centered at 1368 cm−1 which could be ascribed to the E2g symmetric vibration mode due to the in-plane atomic displacement of B and N atoms (see Fig. 1c).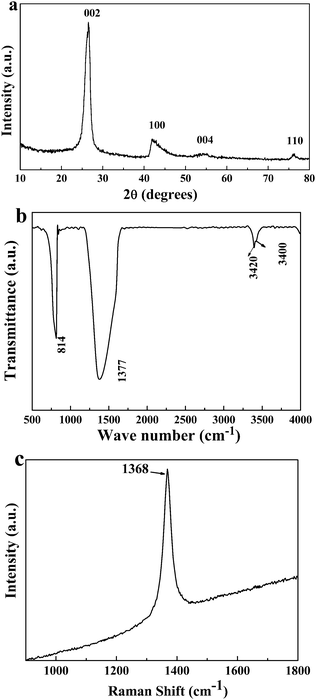 | ||
| Fig. 1 (a) The typical XRD pattern of the sample obtained at 450 °C for 30 h, (b) the FTIR and (c) Raman spectrum of the as-formed sample. | ||
The morphology of the sample was characterized by TEM and HRTEM. Fig. 2a shows a typical TEM image of the as-prepared sample, in which microboxes with well-defined shape can be observed. The sizes of the boxes typically range from 0.5 to 1.4 μm, and the strong contrast between the dark edges and the brighter centre is clearly indicative of their hollow nature. Fig. 2b gives the SEM image of an individual microbox with parallelepiped hexahedral morphology. A typical HRTEM image of the corner of a BN microbox is shown in Fig. 2c, the interspace between two adjacent layers is ∼0.34 nm, which corresponds to that of the bulk h-BN (0.333 nm). The selected area electron diffraction pattern (inset in Fig. 2c) could be indexed to the (002), (100), (004) and (110) planes of h-BN crystallites and the polycrystalline nature of BNMB could be confirmed. The SEM image of the obtained sample is shown in Fig. 2d, which demonstrates that the structural integrity of BNMB is retained after processing. It is worth noting that some microboxes show twisted and disordered facets, which is also displayed in the corresponding TEM image (inset in Fig. 2d). The sample after ultrasonic treatment for five minutes shown in Fig. 2e further implies that these BNMB have hollow structures. Most of BNMB surfaces are cracked to some extent due to the introduction of high intensity ultrasonic vibration.
 | ||
| Fig. 2 (a) A typical TEM image of the as-obtained BNMB. (b) SEM of an enlarged view of a microbox. (c) HRTEM image of a part of one microbox shell. Inset in (c) shows the corresponding SAED pattern of the microbox. (d) Low-magnification SEM and corresponding TEM images of BNMB (inset in d). (e) SEM image of broken hollow microboxes after ultrasonic treatment. | ||
The UV absorption spectrum was used to investigate the optical properties of the sample (Fig. 3a). The absorption band centered at 200 nm could be attributed to the saddle point transition Q2−–Q2− in the band structure of h-BN.36 It is noteworthy that the spectrum shows multiple small split absorption bands near 200 nm, which may be the reflection of phonon–electron coupling.37 Cathodoluminescence (CL) spectroscopy was used to evaluate the luminescence property of the sample. In Fig. 3b, a relatively broad emission peak with a maximum value at 330 nm is observed, which can be assigned to deep levels in the band gap due to intrinsic impurities or structure defects, such as O impurities and nitrogen or boron vacancies that are caused by the curling up h-BN sheets.38 The UV absorption and CL emission properties, similar to previous reports, reveal that BNMB are promising candidates for deep-blue and UV applications such as blue light and UV emitters.
 | ||
| Fig. 3 UV-Vis absorption spectrum (a) and the corresponding CL emission spectrum (b) of the sample measured at room temperature. | ||
A nitrogen cryo-adsorption/desorption isotherm and the corresponding pore size distribution curve of the sample are shown in Fig. 4a. The curve displays an adsorption–desorption hysteresis at a relative pressure of approximately 0.3–1.0, which can be attributed to the formed inhomogeneous mesopores during the aggregation of the nanocrystals. The value of specific surface area of the sample is 86.9 m2 g−1, which is larger than that of the commercial h-BN (∼25.3 m2 g−1), and the average pore size of the sample derived from the desorption branch of the isotherm is 3.97 nm with a narrow distribution (inset in Fig. 4a). The TGA curve of the sample is observed from Fig. 4b. The weight of the product has not changed significantly below 900 °C, while an obvious weight gain is observed between 900–1100 °C for the oxidation of the h-BN to B2O3. Therefore, the high specific surface area, narrow pore size distributions, and excellent thermal stability together with chemical inertness make BNMB a promising catalysis support for catalytic applications.
 | ||
| Fig. 4 (a) Nitrogen adsorption–desorption isotherm of the BNMB, (b) TGA curve of the as-prepared sample under flowing air. | ||
To investigate the formation mechanism of the BNMB, the composition and morphology of the crude product without any post-treatment were studied. The XRD pattern of the crude product (Fig. 5a) can be indexed as KZnF3 (JCPDS card no. 06-0439) and tetragonal ZnF2 (JCPDS card no. 07-0214). Since the as-formed KZnF3 and ZnF2 are sensitive to water in an ambient atmosphere, ZnO can also be detected by XRD. Due to the low diffraction intensity, peaks of boron nitride were not obvious in the pattern. The core–shell structured cubes can be detected by TEM observation of the crude product, as shown in Fig. 5b. The EDX spectrum of these cubes shown in Fig. 5c displays that there are B, N, K, Zn, F and O (originates from the moisture absorbed on their surfaces) elements. We have also probed other cubes, and we confirm that only KZnF3 were covered with h-BN layers. Thus, according to the above complementary analysis, an in situ template route for the formation of BNMB was proposed. During the reaction process, the cube-like KZnF3 particles were firstly formed at a lower temperature. Theoretical calculations show that cubic KZnF3 crystal planes (200), (210) and h-BN (101), (102) exist with small lattice mismatches of about 1.6% and 0.20%, respectively. Like that of ZnS, MgO and Ni,20,39,40 the small lattice mismatch may orient deposition of the h-BN crystals preferentially onto the KZnF3 surface. Finally, the h-BN@ KZnF3 core–shell structure was formed, and a hollow h-BN microbox could be obtained as the crude sample was treated in dilute hydrochloric acid. Based on the above results, the chemical equation can be tentatively described as follows:
| KBH4 + 3Zn + 7NH4F = BNMB + KZnF3 + 2ZnF2 + 7H2 + 6NH3 |
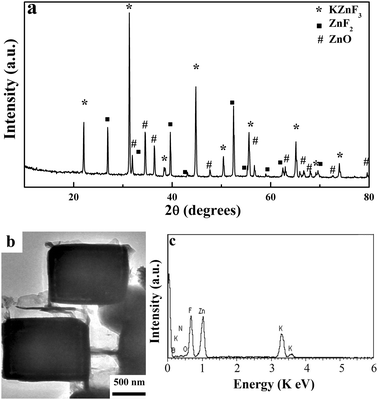 | ||
| Fig. 5 (a) Typical XRD patterns of the crude products without any post treatment. (b) A typical TEM image of the intermediate solid cubes in crude sample and its (c) corresponding EDX spectrum. | ||
 | ||
| Fig. 6 (a) XRD diffraction patterns of the SnO2/BNMB. The inset is the corresponding TEM image. (b) The UV-Vis diffuse reflectance spectra of the SnO2/BNMB, inset shows the plots of (ahν)1/2versus the energy of light (hν). | ||
With the aim of exploring the photocatalytic activity of the SnO2/BNMB, the visible light-driven photocatalytic reaction was evaluated by photodegradation of the MO solution (25 mg L−1). Photocatalytic behaviours of SnO2, SnO2/Al2O3 and Degussa P25 were also measured for comparison. Fig. 7a shows the absorption spectra of MO solution decomposed by SnO2/BNMB under exposure to a 300 W xenon lamp (λ > 420 nm) for various irradiation times. The characteristic absorption of MO at 464 nm had a red-shift to a longer wavelength range during the degradation process, and it completely disappeared after irradiation for 30 min. Meanwhile, a new peak centering around 245 nm was emerged in accordance with a previous study.42 Plots of the removal efficiencies of MO with respect to decomposition time over different catalysts are shown in Fig. 7b. Owing to their relatively large band gap, P25 and pure SnO2 are inactive for the degradation of MO under visible irradiation. The degradation efficiency of MO by SnO2/BNMB reached 92.0% after 30 min irradiation, whereas the MO removal only reached 21.6% in the presence of SnO2/Al2O3. Evidently, supports have significant influences on the catalytic activity of photocatalysts, and BNMB are more effective and supportive for SnO2 to degrade MO in aqueous solutions compared to conventional Al2O3. Several reasons should be responsible for the prominent photocatalytic activity of SnO2/BNMB. First, the absorption edge of the SnO2/BNMB is located in the visible light region, which is a crucial factor that leads to enhanced visible light absorption and converts the excitation light to chemical energy to accelerate photocatalytic oxidation. Second, as a support, BNMB exhibits strong adsorption ability for MO due to its layered structure, hydrophobic property and high surface area, thus MO will concentrate around the loaded SnO2 nanoparticles. The good contact between the MO and SnO2 is beneficial to enhance the photocatalytic efficiency of SnO2/BNMB. Moreover, layered BNMB in contact with SnO2 also suppress the recombination of photogenerated electrons and holes at SnO2/BNMB interfaces, allowing both of them to participate in the photocatalytic reaction. Third, SnO2 particles loaded on BNMB, with improved dispersion and relatively small sizes, help to increase photocatalytic active sites through a great increase in the particles' surface areas. Thus, the SnO2/BNMB successfully combines the catalytic and adsorptive properties of SnO2 and BNMB, which lead to their prominent photoactivity.
 | ||
| Fig. 7 (a) The absorption spectral changes of MO solution over SnO2/BNMB under visible light irradiation (λ > 420 nm), (b) MO photodegradation over SnO2/BNMB, SnO2, SnO2/Al2O3 and P25 with irradiation times. | ||
The mass spectrum (MS) analysis technique has been used to determine the degradation intermediates of MO and preliminary study on the possible mechanism of the photodegradation process. As shown in Fig. 8a, the MO solution before irradiation provides only a single MS signal at m/z = 304 (1.25 × 105 counts) attributed to ionization of the parent MO molecule. After visible light irradiation for 30 min, the absorption peak intensity of m/z = 304 largely decreased (6.05 × 102 counts) and some new peaks appeared at m/z = 249, m/z = 173, m/z = 156, m/z = 121 and m/z = 113 corresponding to intermediates of the MO molecular structure (shown in Fig. 8b). As can be seen, MO has been decomposed but not mineralized completely after visible light irradiation (λ > 420 nm) for 30 min. Based on the m/z values of the intermediates and the structure of MO, the possible degradation pathway for MO is presented in Fig. 8c. Some fragment ions of m/z = 189 and m/z = 143 were not detected, probably because of their corresponding intermediate concentrations were very low or their unstable structure during the degradation of MO. On the basis of previous experimental reports,23,29,43 we consider that free radicals ˙OH, ˙O2– and O2 absorbed on the surface of SnO2/BNMB as oxidation agents could degrade the organic dye through two primary processes of demethylation and hydroxylation. During the initial 30 min of irradiation, MO dye was degraded into lower molecular weight intermediates. As the irradiation time prolonged, these intermediates would finally be degraded to final non-toxic inorganic products.
 | ||
| Fig. 8 Mass spectrum of MO without visible-light irradiation (a) and its degradation intermediates after 30 min of irradiation (b). (c) Proposed photodegradation process on the basis of MS analysis during photodegradation of MO. | ||
4 Conclusions
In conclusion, hexagonal BNMB were prepared from KBH4, NHF4 and Zn powder at 450 °C for 30 h. The as-prepared BNMB with excellent thermal stability, optical and chemical properties, and a high surface area of ∼86.9 m2 g−1 are considered an ideal catalyst support. Thus, SnO2 was loaded using a wet chemistry method on surfaces of BNMB for photocatalytic application. The result indicates that the as-formed SnO2/BNMB has the highest photocatalytic activity compared with common photocatalysts, and its degradation rate is nearly 92% within 30 min towards MO under visible light irradiation (λ > 420 nm). The excellent photocatalytic performance is related to the narrow band gap of SnO2/BNMB, the improved dispersion and relatively small size of loading SnO2 particles, the hydrophobicity, large specific surface area and the layered structure of the BNMB support. Moreover, the MS analysis technique has been used to determine the main degradation intermediates and the possible photodegradation process of an MO molecule. The effective in situ template approach for synthesizing BNMB may be extended to synthesize other box-like inorganic compounds. The excellent photocatalytic performance of SnO2/BNMB indicates that BNMB are a promising support, which should be used as an important guide to further design more useful photocatalytic systems.Acknowledgements
This work was supported by the National Nature Science Found of China (Grant Nos. 20871075 and 20971079), the 973 Project of China (No. 2011CB935901), and the Independent Innovation Foundations of Shandong University (Grant Nos. 2009TS017, 2009JC019).Notes and references
- X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987 CrossRef CAS.
- S. J. Liu, X. X. Wu, B. Hu, J. Y. Gong and S. H. Yu, Cryst. Growth Des., 2009, 9, 1511 CAS.
- M. R. Kim and D. J. Jang, Chem. Commun., 2008, 5218 RSC.
- Y. G. Sun and Y. N. Xia, Science, 2002, 298, 2176 CrossRef CAS.
- Z. M. Peng, H. J. You, J. B. Wu and H. Yang, Nano Lett., 2010, 10, 1492 CrossRef CAS.
- X. D. Su, J. Z. Zhao, H. Bala, Y. C. Zhu, Y. Gao, S. S. Ma and Z. C. Wang, J. Phys. Chem. C, 2007, 111, 14689 CAS.
- S. H. Jiao, L. F. Xu, K. Jiang and D. S. Xu, Adv. Mater., 2006, 18, 1174 CrossRef CAS.
- X. F. Yang, J. X. Fu, C. J. Jin, J. Chen, C. L. Liang, M. M. Wu and W. Z. Zhou, J. Am. Chem. Soc., 2010, 132, 14279 CrossRef CAS.
- T. He, D. R. Chen, X. L. Jiao and Y. L. Wang, Adv. Mater., 2006, 18, 1078 CrossRef CAS.
- Z. Y. Wang, D. Y. Luan, C. M. Li, F. B. Su, S. Madhavi, F. Y. C. Boey and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 16271 CrossRef CAS.
- S. Bastide, D. Duphil, J. P. Borra and C. Lévy-Clément, Adv. Mater., 2006, 18, 106 CrossRef CAS.
- D. F. Liu, S. H. Yang and S. T. Lee, J. Phys. Chem. C, 2008, 112, 7110 CAS.
- S. T. Chen, X. L. Zhang, X. M. Hou, Q. Zhou and W. H. Tan, Cryst. Growth Des., 2010, 10, 1257 CAS.
- C. S. Mei, Z. C. Liu, P. Y. Wen, Z. K. Xie, W. M. Hua and Z. Gao, J. Mater. Chem., 2008, 18, 3496 RSC.
- X. F. Yang, J. X. Fu, C. J. Jin, J. Chen, C. L. Liang, M. M. Wu and W. Z. Zhou, J. Am. Chem. Soc., 2010, 132, 14279 CrossRef CAS.
- R. Haubner, M. Wilhelm, R. Weissenbacher and B. Lux, Struct. Bonding, 2002, 102, 1 CrossRef CAS.
- M. D. Ganji, H. Yazdani and A. Mirnejad, Physica E (Amsterdam), 2010, 42, 2184 CAS.
- X. Chen, P. Wu, M. Rousseas, D. Okawa, Z. Gartner, A. Zettl and C. R. Bertozzi, J. Am. Chem. Soc., 2009, 131, 890 CrossRef CAS.
- M. Bechelany, A. Brioude, P. Stadelmann, S. Bernard, D. Cornu and P. Miele, J. Phys. Chem. C, 2008, 112, 18325 CAS.
- Z. G. Chen, J. Zou, G. Liu, F. Li, Y. Wang, L. Z. Wang, X. L. Yuan, T. Sekiguchi, H. M. Cheng and G. Q. Lu, ACS Nano, 2008, 2, 2183 CrossRef CAS.
- T. Ohashi, Y. T. Wang and S. Shimada, J. Mater. Chem., 2010, 20, 5129 RSC.
- Z. H. Yang, L. Shi, L. Y. Chen, Y. L. Gu, P. J. Cai, A. W. Zhao and Y. T. Qian, Chem. Phys. Lett., 2005, 405, 229 CrossRef CAS.
- H. J. Wang, F. Q. Sun, Y. Zhang, L. S. Li, H. Y. Chen, Q. S. Wu and J. C. Yu, J. Mater. Chem., 2010, 20, 5641 RSC.
- G. Wang, W. Lu, J. H. Li, J. Y. Choi, Y. Jeong, S. Y. Choi, J. B. Park, M. K. Ryu and K. Lee, Small, 2006, 2, 1436 CrossRef CAS.
- J. C. S. Wu, Y. C. Fan and C. A. Lin, Ind. Eng. Chem. Res., 2003, 42, 3225 CrossRef CAS.
- D. Szmigiel, W. Rarog-Pilecka, E. Miskiewicz, E. Maciejewska, Z. Kaszkur, J. W. Sobczak and Z. Kowalczyka, Catal. Lett., 2005, 100, 79 CrossRef CAS.
- G. Postole, A. Gervasini, M. Caldararu, B. Bonnetot and A. Auroux, Appl. Catal., A, 2007, 325, 227 CrossRef CAS.
- J. C. S. Wu, C. Y. Chen and S. D. Lin, Catal. Lett., 2005, 102, 223 CrossRef CAS.
- E. Dvininova, M. Ignata, P. Barvinschi, M. A. Smithersc and E. Popovici, J. Hazard. Mater., 2010, 177, 150 CrossRef.
- H. Yamashita, H. Nose, Y. Kuwahara, Y. Nishida, S. Yuan and K. Mori, Appl. Catal., A, 2008, 350, 164 CrossRef CAS.
- Y. Kuwahara, K. Maki, Y. Matsumura, T. Kamegawa, K. Mori and H. Yamashita, J. Phys. Chem. C, 2009, 113, 1552 CAS.
- M. Shang, W. Z. Wang and L. Zhang, J. Hazard. Mater., 2009, 167, 803 CrossRef CAS.
- C. Y. Zhi, Y. Bando, C. C. Tang and D. Golberg, J. Phys. Chem. B, 2006, 110, 8548 CrossRef CAS.
- M. Chhowalla and G. A. J. Amaratunga, Nature, 2000, 407, 164 CrossRef CAS.
- R. Geick, C. H. Perry and G. Rupprecht, Phys. Rev., 1966, 146, 543 CrossRef CAS.
- J. S. Laurent, R. Arenal, F. Ducastelle, A. Loiseau, M. Cau, B. Attal-Tretout, E. Rosencher and L. Goux-Capes, Phys. Rev. Lett., 2005, 94, 037405 CrossRef.
- B. S. Zou, Y. Zhang, L. Z. Xiao and T. J. Li, J. Appl. Phys., 1993, 73, 4689 CrossRef CAS.
- W. Q. Han, H. G. Yu, C. Y. Zhi, J. B. Wang, Z. Liu, T. Sekiguchi and Y. Bando, Nano Lett., 2008, 8, 491 CrossRef CAS.
- L. C. Wang, L. Q. Xu, C. H. Sun and Y. T. Qian, J. Mater. Chem., 2009, 19, 1989 RSC.
- L. Q. Xu, J. H. Zhan, J. Q. Hu, Y. Bando, X. L. Yuan, T. Sekiguchi, M. Mitome and D. Golberg, Adv. Mater., 2007, 19, 2141 CrossRef CAS.
- J. H. Park and P. M. Woodward, Int. J. Inorg. Mater., 2000, 2, 153 CrossRef CAS.
- M. Sun, D. Z. Li, W. J. Li, Y. B. Chen, Z. X. Chen, Y. H. He and X. Z. Fu, J. Phys. Chem. C, 2008, 112, 18076 CAS.
- S. S. Wu, H. Q. Cao, S. F. Yin, X. W. Liu and X. R. Zhang, J. Phys. Chem. C, 2009, 113, 17893 CAS.
| This journal is © The Royal Society of Chemistry 2011 |