[VO(acac)2] hybrid catalyst: from complex immobilization onto silica nanoparticles to catalytic application in the epoxidation of geraniol†
Clara
Pereira
a,
José F.
Silva‡
a,
André M.
Pereira
b,
João P.
Araújo
b,
Ginesa
Blanco
c,
Jose M.
Pintado
c and
Cristina
Freire
*a
aREQUIMTE, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre s/n, 4169-007 Porto, Portugal. E-mail: acfreire@fc.up.pt; Fax: +351 220402659; Tel: +351 220402590
bIFIMUP and IN – Institute of Nanoscience and Nanotechnology, Departamento de Física e Astronomia, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre s/n, 4169-007 Porto, Portugal
cDepartamento de Ciencia de Materiales e Ingeniería Metalúrgica y Química Inorgánica, Facultad de Ciencias, Universidad de Cádiz, Campus Rio San Pedro, Aptdo. 40, 11510 Puerto Real, Cádiz, Spain
First published on 31st May 2011
Abstract
This work reports the preparation of a novel hybrid nanocatalyst through the immobilization of oxidovanadiumIV acetylacetonate ([VO(acac)2]) onto silica nanoparticles functionalized with 3-aminopropyltriethoxysilane (APTES) and its catalytic application in the allylic epoxidation of geraniol. All the nanomaterials were characterized by chemical analysis, X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FTIR). The parent and amine-functionalized silica nanoparticles were also analyzed by 29Si and 13C solid-state nuclear magnetic resonance and the supported [VO(acac)2] nanomaterial was characterized by magnetic susceptibility measurement. The APTES organosilane was grafted onto the silica nanoparticles with an efficiency of 90%, through mono- and bidentate covalent grafting. The [VO(acac)2] complex was covalently anchored on the surface of the APTES-functionalized silica nanoparticles with 25% of efficiency. Magnetic susceptibility measurements in combination with XPS indicated that the oxidation state of the metal center (VO2+) was preserved upon the immobilization reaction. The catalytic performance of the novel hybrid [VO(acac)2] nanocatalyst was evaluated in the epoxidation of geraniol, at room temperature, using tert-butyl hydroperoxide as an oxygen source. The nanocatalyst presented 100% of substrate conversion, 99% of selectivity towards the 2,3-epoxygeraniol product and was stable upon reuse in further four cycles.
1 Introduction
Over the years, catalysis has been playing a crucial role in the chemical industry with significant economic and environmental impact. Transition metal complexes are of paramount importance in oxidative catalysis for the production of valuable intermediates for organic synthesis such as epoxides.1–3 In particular, oxidovanadiumIV acetylacetonate ([VO(acac)2]) is a very efficient homogeneous catalyst in the epoxidation of allylic alcohols into the corresponding epoxides, using tert-butyl hydroperoxide (t-BuOOH) as an oxidant, presenting very high activity, stereo- and regioselectivity.4–9 Despite the advantages of homogeneous catalysts, their high cost, low chemical and thermal stability and difficulties associated with their separation from the reaction media are major drawbacks towards the goals of Green Chemistry.1,10–13 In this framework, the immobilization of transition metal complexes onto solid supports such as inorganic materials and organic polymers constitutes a potential strategy to overcome these problems,1,10–14 leading to the development of more sustainable catalytic processes.The [VO(acac)2] complex has already been anchored onto several types of bulk materials by covalent bonding (either directly or through a linker between the complex and support)15–18 and by encapsulation,19 for the heterogeneous epoxidation of allylic alcohols. This complex has been immobilized onto amine-functionalized smectite-type clays,15 porous clay heterostructure,16 mesoporous silicas16,17 and activated carbon18 by Schiff condensation between the carbonyl group of the acetylacetonate ligand and the free amine groups previously grafted on the supports surface. It has also been directly anchored onto clays15 and onto a hexagonal mesoporous silica17 by reaction between the complex and the free silanol groups of the support through one of two possible mechanisms: (i) hydrogen bonding between the pseudo π system of the acetylacetonate ligand and the silanol protons of the support or (ii) ligand-exchange with the formation of a covalent bond between the vanadium centre and an oxygen atom from the support.15,20–23 Furthermore, a different immobilization route has been the complex microencapsulation in polystyrene, i.e., the physical envelopment of the complex by a thin polymer film involving interactions between the complex vacant orbitals and the π electrons of the polymer benzene rings.19
Concerning the catalytic performance, the [VO(acac)2] microencapsulated in polystyrene19 and anchored onto amine-functionalized K10-montmorillonite clay15 and activated carbon18 were the most active heterogeneous catalysts in the epoxidation of allylic alcohols, presenting similar substrate conversion as the homogeneous counterpart and good/moderate stability upon recycling/reuse in further cycles without significant metal leaching. Nevertheless, in most cases an increase of the reaction time relative to the homogeneous phase reaction was acknowledged, due to diffusion constraints of the substrate and oxidant to the catalytic active centers.
In this context, the quest for novel recyclable catalysts that combine high substrate conversion/selectivity with low reaction time continues to be a challenging milestone in catalysis.
In the last decades, silica nanoparticles have conquered new horizons in a myriad of research areas including Materials Science and Engineering, Medicine and Biology.24–27 The nanometre size, large surface area to volume ratio and presence of silanol groups on the surface, which allow the incorporation of a wide variety of functionalities, are some of the outstanding features24,25,27 that make silica nanoparticles a promising support for the fabrication of the so-called “quasi-homogeneous” catalysts. Moreover, they can be easily dispersed in several solvents (aqueous and organic) which facilitates the accessibility of the reactants to the catalytic active sites, thus avoiding the diffusion problems typically associated with heterogeneous bulk catalytic systems.28 Despite these potentialities, until now only a few publications have reported the catalytic activity of transition metal complexes anchored onto silica nanoparticles;29–32 for instance, hybrid nanocatalysts containing N-heterocyclic carbene–palladium complexes for Suzuki and Heck coupling reactions,29 chiral RuCl2–diphosphine–diamine complexes for the asymmetric hydrogenation of aromatic ketones30 and Co(III) salen complexes for the hydrolytic kinetic resolution of epichlorohydrin.31 All these nanocatalysts presented remarkable results in terms of activity, selectivity and stability upon reuse.
Very recently, we prepared a [VO(acac)2] magnetic nanocatalyst by immobilization of the complex onto superparamagnetic silica-coated maghemite (γ-Fe2O3) nanoparticles functionalized with amine groups (240 nm particle size).33 In a preliminary study, the magnetic nanocatalyst was tested in the epoxidation of geraniol, leading to similar activity and selectivity as the homogeneous counterpart,33 which highlighted the importance of nanosize supports for the immobilization of the [VO(acac)2] catalyst. Nevertheless, the reaction time was still much higher than that of the homogeneous phase reaction (30 h vs. 15 min), despite the smaller particle size of the nanocatalyst (240 nm). Consequently, the immobilization of this complex onto colloidal nanoparticles with sizes below 100 nm may provide additional advantages namely increasing the surface area to volume ratio and the concentration of catalytic active sites in the reaction medium.
In this context, we report the preparation of a hybrid [VO(acac)2] nanocatalyst through the immobilization of the complex onto silica nanoparticles of approximately 45 nm size which were previously functionalized with 3-aminopropyltriethoxysilane (APTES). The catalytic performance of the supported complex is evaluated in the epoxidation of geraniol, at room temperature, using t-BuOOH as an oxidant and dichloromethane as a solvent, and compared to that of the free complex. We endeavor to examine the influence of the nanometre size support on the complex immobilization efficiency, location of the grafted complex and anchoring mechanism. Additionally insights into the effect of this type of support on the catalyst activity, stability and recyclability will be provided.
2 Experimental
2.1 Materials, reagents and solvents
[VO(acac)2] (95%), APTES (99%), silica nanoparticles (spherical, 99.5% trace metals basis, according to the supplier specifications), dry toluene (99.8%), chlorobenzene (≥99.5%), geraniol (98%) and t-BuOOH solution 5.0–6.0 M in decane were purchased from Aldrich. The dichloromethane solvent was supplied by Romil (HPLC grade) and chloroform (analytical grade) was from Merck. All reagents and solvents were used as received without further purification.2.2 Catalyst preparation
2.3 Physicochemical characterization
Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) studies were performed at “Centro de Materiais da Universidade do Porto” (CEMUP, Porto, Portugal), using a high-resolution environmental scanning electron microscope (FEI Quanta 400 FEG ESEM) equipped with an energy-dispersive X-ray spectrometer (EDAX Genesis X4M). The [VO(acac)2]APTES@SiO2 sample was analyzed as powder both uncoated and coated with a thin gold–palladium film.The amount of silanol groups in the silica nanoparticles was determined through the alkali neutralization method reported in the literature.34 The qualitative identification of free amine groups in APTES@SiO2 was performed by the ninhydrin test;35 the appearance of a blue/purple color indicates the presence of those groups.
Vanadium contents, obtained by inductively coupled plasma emission spectrometry (ICP-AES) and nitrogen elemental analysis (EA) were performed at “Laboratório de Análises do Instituto Superior Técnico” (Lisboa, Portugal).
Magnetic susceptibility experiments were performed in a Quantum Design MPMS instrument with a Reciprocal Sample Option, by measuring the field-cooled magnetization as a function of temperature in the range of 5–300 K with an applied magnetic field of 0.05 T.
X-Ray photoelectron spectroscopy (XPS) was performed in a Kratos Axis Ultra DLD spectrometer using monochromatic Al Kα radiation (1486.6 eV). The powdered samples were pressed into pellets prior to the XPS studies. To correct possible deviations caused by the electric charge of the samples, the calibration of the binding energies was performed by using the C 1s band at 284.6 eV (C–H/C–C) as internal reference. The XPS spectra were deconvoluted with the XPSPEAK 4.1 software, using non-linear least squares fitting routine after a Shirley-type background subtraction. The surface atomic percentages were calculated from the corresponding peak areas and by using the sensitivity factors provided by the manufacturer.
Solid state nuclear magnetic resonance (NMR) spectra of carbon 13 and silicon 29, 13C CPMAS (cross-polarization magic angle spinning) and 29Si CPMAS were recorded at “Departamento de Química, Universidade de Aveiro” (Aveiro, Portugal) on a 9.4 T Bruker Avance 400 spectrometer, at a spinning rate of 7 kHz for carbon and 5 kHz for silicon. 13C CPMAS NMR spectra were recorded with 4.4 μs 1H 90° pulses, 1.5 ms contact time and 5 s recycle delays. 29Si CPMAS NMR spectra were recorded with 4.25 μs 1H 90° pulses, 8 ms contact time and 5 s recycle delays. The 13C and 29Si chemical shifts (δ) were referenced to glycine and caulinite, respectively.
The Fourier transform infrared (FTIR) spectra of the compounds were collected with a Jasco FT/IR-460 Plus spectrophotometer in the 400–4000 cm−1 range, using a resolution of 4 cm−1 and 32 scans. The spectra of the samples were obtained in KBr pellets (Merck, spectroscopic grade) containing 0.6 wt% of silica-based nanomaterial. All samples and KBr were dried at 100 °C overnight before KBr pellets preparation.
GC-FID chromatograms were obtained with a Varian CP-3380 gas chromatograph equipped with a FID detector, using helium as carrier gas and a fused silica Varian Chrompack capillary column CP-Sil 8 CB Low Bleed/MS (30 m × 0.25 mm id; 0.25 μm film thickness). Temperature program for geraniol epoxidation reactions: 40 °C (1 min), 25 °C min−1, 150 °C, 5 °C min−1, 200 °C (1 min); injector temperature, 200 °C; detector temperature, 250 °C. GC-FID was used to determine the catalytic parameters (geraniol conversions, products selectivities and 2,3-epoxygeraniol yields), since this technique provided the same results as those obtained from 1H NMR and 2,3-epoxygeraniol isolated yield (based on the results reported in our previous work16). In this context, the reaction parameters geraniol conversion (Conv.), 2,3-epoxygeraniol selectivity (S2,3-EG), geranial selectivity (Sgeranial) and 2,3-epoxygeraniol yield (η2,3-EG) were calculated as follows, where A stands for chromatographic peak area:
 | (1) |
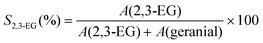 | (2) |
 | (3) |
 | (4) |
 | (5) |
2.4 Catalytic studies
The geraniol epoxidation was carried out at room temperature using 1.00 mmol of geraniol (substrate), 0.50 mmol of chlorobenzene (GC internal standard) and 0.10 g of [VO(acac)2]APTES@SiO2 (catalyst) in 5.00 cm3 of dichloromethane, with continuous stirring. The oxygen source, t-BuOOH, 1.50 mmol, was progressively added to the reaction using a Bioblock Scientific Syringe Pump at a rate of 3.05 cm3 h−1. To monitor the reaction progress, during the catalytic experiment, 0.05 cm3 aliquots were withdrawn from the reaction mixture with a hypodermic syringe, filtered and analyzed by gas chromatography. At the end of the reaction, the heterogeneous nanocatalyst was recovered by centrifugation, refluxed/centrifuged with 50 cm3 of CH2Cl2 for 2 h, dried under vacuum at 120 °C during 2 h, and reused four times. The catalytic reaction was also performed under similar conditions (a) in the homogeneous phase using the same [VO(acac)2] loading as that of the heterogeneous nanomaterial and (b) in the heterogeneous phase using the parent SiO2 and APTES@SiO2 supports.3 Results and discussion
3.1 Morphology and chemical composition
The SEM micrograph of [VO(acac)2]APTES@SiO2 (Fig. 1(A)) shows that the hybrid nanomaterial presents the same morphology as the parent silica nanosupport (∼45 nm particle size, not shown).![(A) SEM micrograph and (B) EDS spectrum of [VO(acac)2]APTES@SiO2 nanomaterial (inset: atomic percentages of C, O, Si and V).](/image/article/2011/CY/c1cy00090j/c1cy00090j-f1.gif) | ||
| Fig. 1 (A) SEM micrograph and (B) EDS spectrum of [VO(acac)2]APTES@SiO2 nanomaterial (inset: atomic percentages of C, O, Si and V). | ||
The EDS analysis (Fig. 1(B)) reveals the presence of carbon (27.9%) and vanadium (0.6%), in addition to oxygen and silicon from the silica framework, providing a first evidence for the complex immobilization onto SiO2; nevertheless, the carbon contribution from the double-sided carbon tape used in the sample preparation cannot be disregarded.
The amount of silanol groups on the surface of the parent SiO2 nanomaterial was determined through the alkali neutralization method. Based on this method, each gram of nanosilica contains 1.51 mmol of OH groups.
The ninhydrin test was performed to detect qualitatively the existence of free amine species on APTES@SiO2 (Fig. S1, ESI†). The colour of the nanomaterial changed from white to blue when the ninhydrin solution was added, confirming the presence of free amine groups. In contrast, the test was negative for the parent SiO2 (no colour change; Fig. S1, ESI†).
The nitrogen and vanadium analyses allowed the determination of the amount of APTES and [VO(acac)2] grafted onto APTES@SiO2 and [VO(acac)2]APTES@SiO2, respectively, and the calculation of their anchoring efficiencies (Table 1).
| Material | N/mmol g−1 | V/μmol g−1 | |||
|---|---|---|---|---|---|
| EA | XPS a | ICP | χ(T) b | XPS a | |
| a Surface contents determined from XPS data in Table 2: mmol N/weight of material = atomic% N/Σ[atomic% (i) × Ar(i)]; μmol V/weight of material = atomic% V/Σ[atomic% (i) × Ar(i)]. b Vanadium bulk content estimated by magnetic susceptibility measurements: μmol V/weight of material = Canc/[Cfree × M([VO(acac)2]). | |||||
| SiO2 | — | — | — | — | — |
| APTES@SiO2 | 1.4 | 2.4 | — | — | — |
| [VO(acac)2]APTES@SiO2 | 1.4 | 2.3 | 295 | 204 | 681 |
| Material | Atomic% | ||||
|---|---|---|---|---|---|
| C 1s | N 1s | O 1s | Si 2p | V 2p | |
| a Determined by the areas of the respective bands in the high-resolution XPS spectra. | |||||
| SiO2 | 4.1 | — | 66.8 | 29.1 | — |
| APTES@SiO2 | 18.9 | 4.5 | 50.8 | 25.8 | — |
| [VO(acac)2]APTES@SiO2 | 21.4 | 4.2 | 50.7 | 22.5 | 1.2 |
The nitrogen loading of APTES@SiO2 is 1.4 mmol g−1, which corresponds to an APTES functionalization efficiency of 56%, estimated by the expression: amount of grafted APTES/amount used in the functionalization reaction × 100. Considering that: (i) the amount of APTES used during the functionalization was higher than the amount of silanol groups on the surface of the parent SiO2, (ii) the amount of grafted APTES is almost similar to the amount of silanol groups on the parent SiO2, and finally (iii) APTES is grafted through bidentate (T2) and monodentate (T1) mechanisms (see NMR data below), the occurrence of a small extent of APTES lateral polymerization may not be disregarded.
In [VO(acac)2]APTES@SiO2 the nitrogen content is similar to that of APTES@SiO2, indicating that no APTES leaching occurred during the complex immobilization. Additionally, the vanadium content is 295 μmol g−1, which corresponds to an anchoring efficiency (amount of anchored complex/amount in original solution × 100) of 25%. If the anchoring efficiency of the metal complex is calculated relative to the amount of grafted APTES (amount of anchored complex/amount of APTES grafted in APTES@SiO2 × 100), the value obtained is 22%. The complex loading in this nanomaterial is higher than those reported for [VO(acac)2] grafted onto amine-functionalized clays, mesoporous silicas (bulk supports) and silica-coated magnetic nanoparticles (nanosupport), which ranged from 35 to 137 μmol g−1; 15–17,33 this improvement is probably due to the nanometre size of SiO2.
The magnetic properties of [VO(acac)2] and of [VO(acac)2]APTES@SiO2 were studied in order to determine the oxidation state of the vanadium centers and estimate the complex loading in [VO(acac)2]APTES@SiO2. In Fig. S2, ESI†, are presented the corresponding temperature-dependent magnetic susceptibility (χ(T)) curves.
The χ(T) curve of the free [VO(acac)2] complex (Fig. S2(a), ESI†) follows the Curie–Weiss law, typical of a paramagnetic state, with an additional small diamagnetic contribution (arising from the acetylacetonate ligands). In order to determine the Curie temperature (ϑP), the χ(T) curve of [VO(acac)2] was fitted using the Curie–Weiss law:
 | (6) |
The χ(T) of the anchored [VO(acac)2] (Fig. S2(b), ESI†) also exhibits a behavior characteristic of a paramagnetic state, revealing that the vanadyl cations remain in the same oxidation state after the immobilization of the complex on the nanosupport. From the Curie–Weiss law (eqn (6)) a Curie constant Canc ≈ 6.65 × 10−5 emu K g−1Oe−1, a diamagnetic component χd ≈ −5.22 × 10−7 emu g−1Oe−1 and a Curie temperature ϑP ≈ −1.2 K for the anchored complex are obtained, which indicate the presence of very weak antiferromagnetic interactions between the VO2+ cations.41
The vanadium content in [VO(acac)2]APTES@SiO2, estimated from the ratio between the Curie–Weiss constants of [VO(acac)2]APTES@SiO2 and [VO(acac)2], is ∼204 μmol g−1 (Table 1), which is comparable to the V loading determined by ICP-AES (295 μmol g−1).
All the nanomaterials were characterized by XPS to gather information regarding the surface chemical composition and the functionalization/immobilization mechanisms. The XPS surface atomic percentages and the core-level binding energies (BEs) are summarized in Tables 2 and 3.
| Material | C 1s | O 1s | N 1s | Si 2p3/2 | Si 2p1/2 | V 2p3/2 | V 2p1/2 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BE a/eV | Areab/% | BE a/eV | Areab/% | BE a/eV | Areab/% | BE a/eV | Areab/% | BE a/eV | Areab/% | BE a/eV | Areab/% | BE a/eV | Areab/% | |
| a The values between brackets refer to the full width at half-maximum of the bands. b Area of each component relative to the total core-level peak area. | ||||||||||||||
| SiO2 | 284.6 (1.7) | 67.2 | 532.7 (1.6) | 100 | — | — | 103.4 (1.4) | 66.7 | 104.0 (1.4) | 33.3 | — | — | — | — |
| 286.3 (1.7) | 21.8 | |||||||||||||
| 288.7 (1.7) | 11.0 | |||||||||||||
| APTES@SiO2 | 284.6 (1.5) | 45.7 | 532.4 (1.5) | 100 | 399.5 (1.9) | 89.1 | 103.1 (1.5) | 66.7 | 103.7 (1.5) | 33.3 | — | — | — | — |
| 285.4 (1.5) | 28.8 | 401.6 (1.9) | 10.9 | |||||||||||
| 286.2 (1.5) | 19.5 | |||||||||||||
| 288.0 (1.5) | 6.0 | |||||||||||||
| [VO(acac)2] APTES@SiO2 | 284.6 (1.6) | 41.9 | 530.3 (1.7) | 9.5 | 399.9 (2.2) | 94.8 | 103.0 (1.7) | 66.7 | 103.6 (1.7) | 33.3 | 516.5 (2.5) | 73.6 | 523.9 (2.5) | 26.4 |
| 285.5 (1.6) | 27.7 | 532.3 (1.7) | 90.5 | 402.1 (2.2) | 5.2 | |||||||||
| 286.4 (1.6) | 17.0 | |||||||||||||
| 288.2 (1.6) | 11.4 | |||||||||||||
| 292.0 (1.6) | 2.0 | |||||||||||||
| [VO(acac)2] | 284.6 (1.7) | 70.7 | 530.7 (1.9) | 100 | — | — | — | — | — | — | 517.1 (2.2) | 73.7 | 524.5 (2.2) | 26.3 |
| 286.9 (1.7) | 27.4 | |||||||||||||
| 289.4 (1.7) | 1.9 | |||||||||||||
The parent SiO2 nanomaterial is mainly composed of oxygen and silicon (Table 2) in the expected O/Si ratio. A small amount of carbon is also present probably due to some organic impurities. In the O 1s and Si 2p3/2 high-resolution XPS spectra are detected bands with BEs of 532.7 and 103.4 eV, respectively (Table 3), which are characteristic of oxygen and silicon in a silica framework.16,33,42
The functionalization of SiO2 with APTES (APTES@SiO2 sample) leads to the appearance of nitrogen as well as to the increase of the carbon surface content and to the decrease of the oxygen and silicon atomic percentages. The N 1s high-resolution spectrum exhibits a peak at 399.5 eV, which is assigned to free amine groups and confirms the grafting of the organosilane on the surface of SiO2,15,16,33,43–46 in accordance with the data from EA and the ninhydrin test; the small shoulder around 401.6 eV is probably associated with some protonated amine species.16,44–46 Additionally, the bands in the O 1s and Si 2p regions are shifted to lower BEs, suggesting that the organosilane was anchored onto the surface of SiO2 by reaction between the APTES ethoxy moieties and the silanol groups from the silica surface.15,16,33,43 The C 1s spectrum exhibits three main components: the bands at 284.6 and 285.4 eV are assigned to C–H/C–C and C–N bonds from the grafted APTES and the band at 286.2 eV is related to C–O bonds probably from unreacted ethoxy groups of the organosilane.42,47
Upon the anchorage of [VO(acac)2] onto APTES@SiO2, a new peak is detected in the V 2p3/2 region with a BE (516.5 eV) characteristic of vanadyl (VO2+) ions, in accordance with magnetic susceptibility measurements, thus confirming the successful immobilization of the complex. Furthermore, the BE of the V 2p3/2 peak is lower than that of the free complex (517.1 eV), indicating a change in the chemical environment of the vanadium centre, which can be interpreted as a consequence of covalent immobilization of the complex to the APTES@SiO2 support. Similarly, both N 1s components are slightly shifted to higher BEs also revealing a change in the nitrogen chemical environment. These features, combined with the high APTES loading which prevents the direct interaction between the complex and the silanol groups, suggest that [VO(acac)2] is covalently bonded to the nanosupport surface through the grafted APTES. There are two major mechanisms for the covalent immobilization of the complex onto the support via APTES spacers (Scheme 1): (i) axial coordination of –NH2 to the vanadium center, or (ii) Schiff condensation between the NH2 group from the grafted APTES and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the acac ligand, leading to the formation of an imine bond coordinated to the metal center: this latter mechanism was proposed in our previous works on the immobilization of [VO(acac)2] onto APTES-functionalized bulk materials (clays, porous clay heterostructure, mesoporous silicas and activated carbon) and silica-coated magnetic nanoparticles.15,16,18,33 However, XPS data per se cannot discriminate unambiguously the two anchoring mechanisms, since both pathways can lead to similar shifts of the V 2p3/2 and N 1s BEs.
O group of the acac ligand, leading to the formation of an imine bond coordinated to the metal center: this latter mechanism was proposed in our previous works on the immobilization of [VO(acac)2] onto APTES-functionalized bulk materials (clays, porous clay heterostructure, mesoporous silicas and activated carbon) and silica-coated magnetic nanoparticles.15,16,18,33 However, XPS data per se cannot discriminate unambiguously the two anchoring mechanisms, since both pathways can lead to similar shifts of the V 2p3/2 and N 1s BEs.
 | ||
| Scheme 1 Schematic representation (not drawn to scale) of complex immobilization onto silica nanoparticles. | ||
The C 1s high-resolution spectrum exhibits four peaks assigned to C–H/C–C (284.6 eV), C–N (285.5 eV), C–O (286.4 eV) and CN/CO (288.2 eV) bonds from the anchored complex and APTES and a weak shoulder at 292.0 eV ascribed to the π–π* shake-up satellite from the π system of the chelate rings.42,48,49
In the O 1s region, besides the band from lattice oxygen, an additional peak at 530.3 eV is observed, which may be assigned to the oxygen-based groups from the anchored complex (O–V/OV and OC bonds).42,50,51 It is also noteworthy that the C 1s and O 1s peaks related to the free [VO(acac)2] complex show a shift to higher and lower BEs, respectively, upon its immobilization onto the nanomaterial.
The comparison between the surface and bulk contents, for N and V elements, provides information concerning the location of the grafted APTES and [VO(acac)2] on the support, respectively. In APTES@SiO2, the nitrogen surface loading determined by XPS is higher than the bulk value obtained by EA (Table 1), revealing that APTES functionalities are preferentially located on the most external surface of the nanomaterial. Similarly, in the [VO(acac)2]APTES@SiO2 nanomaterial the vanadium surface content (681 μmol g−1) is higher than the bulk value (295 μmol g−1), pointing out that [VO(acac)2] is anchored onto the most external surface of the nanosupport. This is an indirect indication of the complex immobilization via APTES spacers, since these groups are also located on the SiO2 outer surface.
3.2 NMR and FTIR studies
29Si and 13C solid-state NMR experiments were performed to unveil the grafting mechanism involved in the functionalization of SiO2 with APTES (Fig. 2). | ||
| Fig. 2 (a) 29Si and (b) 13C CPMAS solid state NMR spectra of SiO2 and APTES@SiO2 and peaks assignments. | ||
The 29Si CPMAS spectrum of SiO2 (Fig. 2(a)) exhibits three resonance peaks at δ = −109, −100 and −91 ppm, which correspond to Q4 (fully condensed silica units), Q3 (terminal silanol groups) and Q2 (geminal silanol groups) silicon sites, respectively, where Qn = (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)nSi(OH)4−n, with n = 2–4.23,52–54
SiO)nSi(OH)4−n, with n = 2–4.23,52–54
The grafting of APTES onto SiO2 (APTES@SiO2, Fig. 2(a)) leads to a significant reduction of the intensity of the Q3 and Q2 resonances relative to that of Q4, to the broadening of the Q4 resonance peak and to the appearance of a broad band around −52 ppm assigned to T2 and T1silicon environments (Tm = (SiO)mSi(OH)3−mR, where m = 1–2 and R = –(CH2)3NH2).23,52–54 These features indicate that the organosilane moieties were covalently anchored to the silica backbone through the reaction between the terminal and geminal surface silanol groups of SiO2 and two or one ethoxy groups of APTES (bidentate and monodentate covalent grafting).
The 13C CPMAS NMR spectrum of APTES@SiO2 (Fig. 2(b)) exhibits three resonance peaks at δ = 9.9, 21.8 and 43.0 ppm, which are associated to the three carbon atoms from 3-aminopropyl groups, C1, C2 and C3, respectively,44,47,52–54 confirming that the grafted APTES functionalities preserved their structural integrity. The broadening of the C2 peak at δ = 21.8 ppm suggests the presence of a small fraction of protonated amine groups,55 in accordance with XPS. The low intensity peaks at δ = 16.8 and 58.1 ppm are assigned to a small amount of unreacted ethoxy groups from APTES (C4 and C5 carbon atoms in Fig. 2(b)),47,53,54 since the organosilane is covalently attached to the silica network through 1- or 2-fold covalent grafting.
The silica-based nanomaterials were characterized by FTIR spectroscopy and the corresponding spectra are depicted in Fig. 3.
![FTIR spectra of samples (A) SiO2, (B) APTES@SiO2, (C) [VO(acac)2]APTES@SiO2 and (D) [VO(acac)2], in the (a) 4000–400 cm−1 range and (b) amplified 1750–650 cm−1 region. The red and blue lines highlight the characteristic peaks of the grafted APTES and anchored [VO(acac)2] respectively.](/image/article/2011/CY/c1cy00090j/c1cy00090j-f3.gif) | ||
| Fig. 3 FTIR spectra of samples (A) SiO2, (B) APTES@SiO2, (C) [VO(acac)2]APTES@SiO2 and (D) [VO(acac)2], in the (a) 4000–400 cm−1 range and (b) amplified 1750–650 cm−1 region. The red and blue lines highlight the characteristic peaks of the grafted APTES and anchored [VO(acac)2] respectively. | ||
The FTIR spectrum of the SiO2 sample exhibits the characteristic bands of the silica framework related to Si–O–Si asymmetric stretching (1090 cm−1 with a shoulder around 1220 cm−1), Si–O stretching of Si–OH and Si–O−groups on the nanoparticles surface (948 cm−1), Si–O–Si symmetric stretching (803 cm−1) and Si–O–Si bending vibrations (469 cm−1).16,33,56–59 Additionally, the spectrum presents a band at 1629 cm−1 associated with H–O–H bending vibrations of physically adsorbed water and a broad band centered around 3344 cm−1 due to O–H stretching vibrations of hydrogen-bonded surface silanol groups and physically adsorbed water; the weak shoulders around 3735 and 3650 cm−1 are ascribed to isolated and internal silanol groups, respectively.16,33,56,60
The post-grafting of APTES onto SiO2 is confirmed by the appearance of several new peaks (with low intensity) in all the frequency range, namely in the ranges of 2973–2880 cm−1 and 1472–1389 cm−1 due to C–H stretching and bending vibrations, respectively, and in the range of 1595–1541 cm−1 and at 695 cm−1 assigned to N–H bending vibrations of the aminopropyl groups.15,16,44,45,57,61 Furthermore, ongoing from SiO2 to APTES@SiO2, a significant reduction of the intensity of the O–H stretching and bending vibrations bands (∼3344 and 1629 cm−1, respectively) and of the peak from Si–OH stretching modes (at 948 cm−1) is observed which sustains the grafting mechanism illustrated in Scheme 1, catalyzed by the physisorbed water, in accordance with XPS and 29Si CPMAS NMR results and with the literature.15,16,33,43
The IR bands related to the anchored complex are very weak when compared to those associated with the support and grafted APTES, due to the low complex loading. Nevertheless, the immobilization of [VO(acac)2] onto APTES@SiO2 leads to changes in the C–H stretching vibration region (2979–2880 cm−1) which indirectly indicate the presence of [VO(acac)2] in the nanomaterial, since the free complex exhibits bands in this range.16,62 In the low energy region, the most prominent features are the appearance of an asymmetric and broad band centered around 1653 cm−1 and a peak at 1447 cm−1 that can be unambiguously assigned to the immobilized complex since they are different from those of the free complex and are not detected in the APTES@SiO2 spectrum.62,63 These unique bands are not sufficient to make reliable assignments of the type of vibrations/functionalities involved, preventing the clear discrimination between the two covalent binding mechanisms (i) and (ii) depicted in Scheme 1. In one previous work, we performed theoretical studies at the DFT level to predict the IR spectra of the model compounds representing the two different immobilized V species.16 In that work, the theoretical calculations showed that the CN stretching vibration coupled with the vibration of the acac chelate ring occurred, in the IR spectrum of the complex immobilized through mechanism (ii), at higher wavenumber (1634 cm−1) than the CO/C–O stretching vibrations of the acac chelate ring (1570 cm−1) in the IR spectrum of the complex immobilized by mechanism (i). Therefore, the band at 1653 cm−1 detected in the experimental FTIR spectrum of [VO(acac)2]APTES@SiO2 (Fig. 3) can be tentatively assigned to CN stretching vibrations associated to the formation of an imine bond, suggesting that the complex may be anchored through mechanism (ii). But, as stated before, a full discrimination of the two anchoring mechanisms requires more experimental support.
3.3 Catalytic epoxidation of geraniol
The [VO(acac)2]APTES@SiO2 nanomaterial was tested as a catalyst in the epoxidation of geraniol, at room temperature, in dichloromethane, using t-BuOOH as an oxidant and the results are summarized in Table 4. Data from the homogeneous phase reaction and from the blank experiments (with the parent SiO2 and APTES@SiO2 nanosupports) under similar conditions are also included.
| Sample | Run | t/h | Conv.b/% | Sc/% | 2,3-EG yieldd/% | |
|---|---|---|---|---|---|---|
| 2,3-EG | Geranial | |||||
| a Reaction conditions: 1.00 mmol of geraniol, 0.50 mmol of chlorobenzene (internal standard), 0.10 g of heterogeneous material, 1.50 mmol of t-BuOOH; solvent: dichloromethane. The SiO2 and APTES@SiO2 parent materials led to 0% of geraniol conversion after 2 h. b Based on geraniol consumption. c Products selectivity: 2,3-EG = 2,3-epoxygeraniol. d Determined by GC-FID using chlorobenzene as internal standard and isolated 2,3-EG product. e Homogeneous phase reaction using the same V loading as [VO(acac)2]APTES@SiO2. | ||||||
| [VO(acac)2]APTES@SiO2 | 1st | 2 | 100 | 99 | 1 | 98 |
| 2nd | 2 | 100 | 99 | 1 | 98 | |
| 3rd | 2 | 100 | 98 | 2 | 98 | |
| 4th | 2 | 100 | 98 | 2 | 98 | |
| 5th | 2 | 100 | 98 | 2 | 98 | |
| [VO(acac)2]e | 1st | 0.25 | 100 | 100 | 0 | 100 |
The [VO(acac)2]APTES@SiO2 hybrid nanocatalyst is very active and regioselective towards the 2,3-epoxygeraniol product, leading to 100% of geraniol conversion and 98% of 2,3-epoxygeraniol yield. Furthermore, it exhibits similar activity and regioselectivity as the homogeneous counterpart (100% of geraniol conversion and of 2,3-epoxygeraniol selectivity/yield), albeit an increase in the reaction time from 15 min to 2 h, which may be in part attributed to the modification of the vanadium coordination sphere upon the complex immobilization. A small amount of geranial (1–2%) was also formed.16,33
The lack of activity of the parent nanosupports SiO2 and APTES@SiO2 (0% of geraniol conversion) confirms that the catalytic efficiency of [VO(acac)2]APTES@SiO2 is due to the anchored complex.
The [VO(acac)2]APTES@SiO2 nanomaterial was recycled and reused in further four cycles, preserving its catalytic performance, in terms of reaction time, substrate conversion and 2,3-epoxygeraniol selectivity. Nevertheless, a very small amount of vanadium was detected (by AAS) in the filtered reaction medium after each catalytic cycle, which decreased from cycle to cycle–2.8, 1.9, 1.5, 1.2 and 1.1 μmol for the 1st, 2nd, 3rd, 4th and 5th cycle respectively. These values correspond to 9.5, 6.4, 5.0, 3.9 and 3.8% of leaching and to a total leaching percentage of 28.6. After the five consecutive cycles, the nanocatalyst was also characterized by chemical analyses and the N and V bulk contents were 1.1 mmol g−1 and 216 μmol g−1, respectively, corresponding to leaching percentages of 16 and 27%. The latter value is almost equal to that obtained from the sum of the partial leaching percentages. As neither the reaction time nor the substrate conversion of the [VO(acac)2]APTES@SiO2 nanocatalyst decreased in the consecutive cycles, this may be taken as evidence that the homogeneous component (due to the leached V species) is not significant for the global catalytic activity, although it may exist.
The novel hybrid nanocatalyst prepared herein presents promising results when compared to [VO(acac)2] anchored onto bulk materials (clays, porous clay heterostructure and mesoporous silicas), especially in terms of reaction time (2 h vs. 48 h) and geraniol conversion (100% vs. 34–100%).15–17 The decrease of the reaction time can be assigned to the nanometre size of the support which improves the dispersion of the catalytic active sites in the reaction medium and leads to a higher complex loading.
In fact, very recently, this complex was anchored onto magnetic silica-coated γ-Fe2O3 nanoparticles with ∼240 nm size and the preliminary test of its catalytic performance (100% of geraniol conversion and 96% of 2,3-epoxygeraniol yield) highlighted the potentialities of designing hybrid nanocatalysts for epoxidation reactions.33 However, the reaction time (30 h), although being lower than those of the bulk counterparts, was still significantly higher than the value observed in the homogeneous reaction (15 min). The main differences between the silica-coated magnetic nanocatalyst and the one prepared in this work are the particles size, ∼240 vs. ∼45 nm, and the amount of anchored complex, 35 vs. 295 μmol g−1. From our point of view, the higher complex loading in [VO(acac)2]APTES@SiO2 may be the factor that justifies the significant reduction of the reaction time to 2 h.
4 Conclusions
The [VO(acac)2] complex was covalently anchored onto the surface of APTES-functionalized silica nanoparticles. The novel [VO(acac)2] hybrid nanocatalyst was very active and regioselective in the epoxidation of geraniol at room temperature using t-BuOOH as an oxidant, with 100% of substrate conversion and 98% of 2,3-epoxygeraniol yield after only 2 h, which are comparable to the values obtained with the homogeneous counterpart (100% of geraniol conversion and epoxide yield after 15 min). Furthermore, it could be recycled and reused four times, with similar catalytic activity and regioselectivity. The improved catalytic performance of this hybrid nanocatalyst relative to bulk [VO(acac)2] heterogeneous catalysts, mostly in terms of reaction time, was assigned to the reduced particles size of the support (average particles size below 100 nm) which allowed the immobilization of a higher amount of complex and enhanced the dispersion of the catalytic active sites in the reaction medium, thus improving their accessibility to the substrate and oxidant species.Hence, the immobilization of transition metal complexes onto colloidal nanoparticles with sub-100 nm size opens new perspectives for the design of eco-friendly hybrid catalysts that can bridge the gap between homogeneous and heterogeneous catalysis.
Acknowledgements
This work was funded by Fundação para a Ciência e a Tecnologia (FCT) and FEDER, through projects refs. PTDC/CTM/65718/2006 and PTDC/QUI-QUI/105304/2008 and the Portuguese-Spanish Bilateral Cooperation through Projects REF E-72/10 (Portugal) and PT2009-0126 (Spain). C.P. and A.M.P. thank FCT for their grants.References
- Q.-H. Xia, H.-Q. Ge, C.-P. Ye, Z.-M. Liu and K.-X. Su, Chem. Rev., 2005, 105, 1603–1662 CrossRef CAS.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef CAS.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS.
- K. B. Sharpless and R. C. Michaelson, J. Am. Chem. Soc., 1973, 95, 6136–6137 CrossRef CAS.
- T. Itoh, K. Jitsukawa, K. Kaneda and S. Teranishi, J. Am. Chem. Soc., 1979, 101, 159–169 CrossRef CAS.
- B. E. Rossiter, T. R. Verhoeven and K. B. Sharpless, Tetrahedron Lett., 1979, 20, 4733–4736 CrossRef.
- V. Conte, F. Di Furia and G. Licini, Appl. Catal., A, 1997, 157, 335–361 CrossRef CAS.
- A. G. J. Ligtenbarg, R. Hage and B. L. Feringa, Coord. Chem. Rev., 2003, 237, 89–101 CrossRef CAS.
- C. Bolm, Coord. Chem. Rev., 2003, 237, 245–256 CrossRef CAS.
- M. H. Valkenberg and W. F. Hölderich, Catal. Rev. Sci. Eng., 2002, 44, 321–374 CrossRef CAS.
- A. Corma and H. Garcia, Adv. Synth. Catal., 2006, 348, 1391–1412 CrossRef CAS.
- J. M. Fraile, J. I. García and J. A. Mayoral, Chem. Rev., 2009, 109, 360–417 CrossRef CAS.
- A. Zamboulis, N. Moitra, J. J. E. Moreau, X. Cattoën and M. W. C. Man, J. Mater. Chem., 2010, 20, 9322–9338 RSC.
- A. F. Trindade, P. M. P. Gois and C. A. M. Afonso, Chem. Rev., 2009, 109, 418–514 CrossRef CAS.
- C. Pereira, A. R. Silva, A. P. Carvalho, J. Pires and C. Freire, J. Mol. Catal. A: Chem., 2008, 283, 5–14 CrossRef CAS.
- C. Pereira, K. Biernacki, S. L. H. Rebelo, A. L. Magalhães, A. P. Carvalho, J. Pires and C. Freire, J. Mol. Catal. A: Chem., 2009, 312, 53–64 CrossRef CAS.
- B. Jarrais, C. Pereira, A. R. Silva, A. P. Carvalho, J. Pires and C. Freire, Polyhedron, 2009, 28, 994–1000 CrossRef CAS.
- B. Jarrais, A. R. Silva and C. Freire, Eur. J. Inorg. Chem., 2005, 4582–4589 CrossRef CAS.
- A. Lattanzi and N. E. Leadbeater, Org. Lett., 2002, 4, 1519–1521 CrossRef CAS.
- M. Baltes, O. Collart, P. Van der Voort and E. F. Vansant, Langmuir, 1999, 15, 5841–5845 CrossRef CAS.
- P. Kústrowski, L. Chmielarz, R. Dziembaj, P. Cool and E. F. Vansant, J. Phys. Chem. B, 2005, 109, 11552–11558 CrossRef CAS.
- A.-M. Hanu, S. Liu, V. Meynen, P. Cool, E. Popovici and E. F. Vansant, Microporous Mesoporous Mater., 2006, 95, 31–38 Search PubMed.
- S. Shylesh and A. P. Singh, J. Catal., 2006, 244, 52–64 CrossRef CAS.
- H. E. Bergna and W. O. Roberts, Colloidal Silica: Fundamentals and Applications, CRC Press, Taylor & Francis, Boca Raton, USA, 2006 Search PubMed.
- (a) I. I. Slowing, B. G. Trewyn, S. Giri and V. S.-Y. Lin, Adv. Funct. Mater., 2007, 17, 1225–1236 CrossRef CAS; (b) I. I. Slowing, J. L. Vivero-Escoto, B. G. Trewyn and V. S.-Y. Lin, J. Mater. Chem., 2010, 20, 7924–7937 RSC.
- M. Pagliaro, R. Ciriminna and G. Palmisano, J. Mater. Chem., 2009, 19, 3116–3126 RSC.
- J. M. Rosenholm, C. Sahlgren and M. Lindén, Nanoscale, 2010, 2, 1870–1883 RSC.
- V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743–754 RSC.
- S. Tandukar and A. Sen, J. Mol. Catal. A: Chem., 2007, 268, 112–119 CrossRef CAS.
- D. J. Mihalcik and W. Lin, Angew. Chem., Int. Ed., 2008, 47, 6229–6232 CrossRef CAS.
- C. S. Gill, K. Venkatasubbaiah, N. T. S. Phan, M. Weck and C. W. Jones, Chem.–Eur. J., 2008, 14, 7306–7313 CrossRef CAS.
- A. Kr. Ganai, R. Bhardwaj, S. Hotha, S. S. Gupta and B. L. V. Prasad, New J. Chem., 2010, 34, 2662–2670 RSC.
- C. Pereira, A. M. Pereira, P. Quaresma, P. B. Tavares, E. Pereira, J. P. Araújo and C. Freire, Dalton Trans., 2010, 39, 2842–2854 RSC.
- (a) I. A. Rahman, P. Vejayakumaran, C. S. Sipaut, J. Ismail, M. A. Bakar, R. Adnan and C. K. Chee, Colloids Surf., A, 2007, 294, 102–110 Search PubMed; (b) S. Yuasa, M. Okabayashi, H. Ohno, K. Suzuki and K. Kusumoto, US Pat., 4764497, 1988 Search PubMed.
- (a) S. Moore and W. H. Stein, J. Biol. Chem., 1954, 211, 907–913 CAS; (b) H. F. Hoefnagels, D. Wu, G. de With and W. Ming, Langmuir, 2007, 23, 13158–13163 CrossRef CAS.
- K. Dey, R. K. Maiti and S. K. Sen, Inorg. Chim. Acta, 1976, 20, 197–202 Search PubMed.
- I. Correia, J. Costa Pessoa, M. T. Duarte, M. F. M. da Piedade, T. Jackush, T. Kiss, M. M. C. A. Castro, C. F. G. C. Geraldes and F. Avecilla, Eur. J. Inorg. Chem., 2005, 732–744 CrossRef CAS.
- N. E. Gruhn, L. J. Michelsen and B. L. Westcott, Inorg. Chem., 2002, 41, 5907–5911 Search PubMed.
- S. Di Bella, G. Lanza, A. Gulino and I. Fragalà, Inorg. Chem., 1996, 35, 3885–3890 CrossRef CAS.
- J. Costa Pessoa, I. Cavaco, I. Correia, M. T. Duarte, R. D. Gillard, R. T. Henriques, F. J. Higes, C. Madeira and I. Tomaz, Inorg. Chim. Acta, 1999, 293, 1–11 CrossRef CAS.
- M. A. K. Ahmed, H. Fjellvåg, A. Kjekshus and B. Klewe, Z. Anorg. Allg. Chem., 2004, 630, 2311–2318 Search PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, ed. J. Chastain and R. C. King Jr., Physical Electronics, Inc., Minnesota, USA, 1995 Search PubMed.
- C. Pereira, S. Patrício, A. R. Silva, A. L. Magalhães, A. P. Carvalho, J. Pires and C. Freire, J. Colloid Interface Sci., 2007, 316, 570–579 CrossRef CAS.
- A. S. M. Chong and X. S. Zhao, J. Phys. Chem. B, 2003, 107, 12650–12657 CrossRef CAS.
- S.-W. Song, K. Hidajat and S. Kawi, Langmuir, 2005, 21, 9568–9575 CrossRef CAS.
- K. M. R. Kallury, P. M. MacDonald and M. Thompson, Langmuir, 1994, 10, 492–499 CrossRef CAS.
- S. B. Hartono, S. Z. Qiao, J. Liu, K. Jack, B. P. Ladewig, Z. Hao and G. Q. M. Lu, J. Phys. Chem. C, 2010, 114, 8353–8362 CrossRef CAS.
- S. Kundu, Y. Wang, W. Xia and M. Muhler, J. Phys. Chem. C, 2008, 112, 16869–16878 CrossRef CAS.
- L. Britcher, H. Rahiala, K. Hakala, P. Mikkola and J. B. Rosenholm, Chem. Mater., 2004, 16, 5713–5720 Search PubMed.
- R. Molina, M. A. Centeno and G. Poncelet, J. Phys. Chem. B, 1999, 103, 6036–6046 Search PubMed.
- A. R. Silva, M. Martins, M. M. A. Freitas, J. L. Figueiredo, C. Freire and B. de Castro, Eur. J. Inorg. Chem., 2004, 2027–2035 CrossRef CAS.
- T. Yokoi, H. Yoshitake and T. Tatsumi, J. Mater. Chem., 2004, 14, 951–957 RSC.
- M. A. B. Meador, E. F. Fabrizio, F. Ilhan, A. Dass, G. Zhang, P. Vassilaras, J. C. Johnston and N. Leventis, Chem. Mater., 2005, 17, 1085–1098 CrossRef CAS.
- G. S. Caravajal, D. E. Leyden, G. R. Quinting and G. E. Maciel, Anal. Chem., 1988, 60, 1776–1786 CrossRef CAS.
- Z. Luan, J. A. Fournier, J. B. Wooten and D. E. Miser, Microporous Mesoporous Mater., 2005, 83, 150–158 CrossRef CAS.
- A. Bertoluzza, C. Fagnano, M. A. Morelli, V. Gottardi and M. Guglielmi, J. Non-Cryst. Solids, 1982, 48, 117–128 CAS.
- R. Scaffaro, L. Botta, G. L. Re, R. Bertani, R. Milani and A. Sassi, J. Mater. Chem., 2011, 21, 3849–3857 RSC.
- P. Innocenzi, P. Falcaro, D. Grosso and F. Babonneau, J. Phys. Chem. B, 2003, 107, 4711–4717 CrossRef CAS.
- R. Al-Oweini and H. El-Rassy, J. Mol. Struct., 2009, 919, 140–145 CrossRef CAS.
- J.-P. Gallas, J.-M. Goupil, A. Vimont, J.-C. Lavalley, B. Gil, J.-P. Gilson and O. Miserque, Langmuir, 2009, 25, 5825–5834 CrossRef CAS.
- X. Wang, K. S. K. Lin, J. C. C. Chan and S. Cheng, J. Phys. Chem. B, 2005, 109, 1763–1769 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, Inc., New York, 1997 Search PubMed.
- P. Van der Voort, I. V. Babitch, P. J. Grobet, A. A. Verberckmoes and E. F. Vansant, J. Chem. Soc., Faraday Trans., 1996, 92, 3635–3642 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Ninhydrin test for SiO2 and APTES@SiO2 nanomaterials and temperature-dependent magnetic susceptibility curves of [VO(acac)2] and [VO(acac)2]APTES@SiO2. See DOI: 10.1039/c1cy00090j |
| ‡ Present address: CeNTI-Centro de Nanotecnologia e Materiais Técnicos, Funcionais e Inteligentes, Rua Fernando Mesquita 2785, 4760-034 Vila Nova de Famalicão, Portugal. |
| This journal is © The Royal Society of Chemistry 2011 |