Foam, fleece and honeycomb: catalytically active coatings from colloidally prepared nanoparticles
Patrick
Sonström
a,
Birte
Halbach
a,
Sonia
Tambou Djakpou
a,
Beate
Ritz
b,
Kirsten
Ahrenstorf
b,
Georg
Grathwohl
c,
Horst
Weller
b and
Marcus
Bäumer
*a
aInstitute of Applied and Physical Chemistry, University of Bremen, Leobener Str. NW2, 28359 Bremen, Germany. E-mail: mbaeumer@uni-bremen.de; Fax: +49 421-21863188
bInstitute of Physical Chemistry, University of Hamburg, Grindelallee 117, 20146 Hamburg, Germany
cCeramic Materials and Components, University of Bremen, Am Biologischen Garten 2/IW3, 28359 Bremen, Germany
First published on 15th June 2011
Abstract
Although monolithic catalysts offer distinct advantages such as low pressure drops and easy catalyst separation compared to tube bundle reactors filled with pellets and are for instance widely used for automobile exhaust treatment, the coating of the monolithic structures with the catalytically active phase is often complex and recipes need to be established empirically. In contrast to traditional methods, such as dip-coating or impregnation of the monolith, where the active nanoparticles are formed during the preparation on an oxidic washcoat usually used and deposited simultaneously or in a previous step, we demonstrate in this study that alternatively preformed colloidally synthesized nanoparticles can be employed to obtain homogeneous coatings with or without a washcoat. In this way, one can take advantage of the far-reaching possibilities of colloidal methods to control the structure and size of the nanoparticles and also to tune and optimize their binding to the monolithic surface. For cases where beneficial metal–support interactions between the nanoparticles and a washcoat improve the catalytic properties we demonstrate that colloidally prepared nanoparticles can be directly mixed with a washcoat slurry and successfully deposited on monolithic supports. Turnover frequencies comparable to the corresponding powder catalysts could be reached. In a second approach, we present here a facile method to directly coat three different monolithic supports (cordierite honeycomb, Al2O3 foam and Nickel fleece) with preformed Pt nanoparticles in the presence and absence of organic ligands. In order to realize high metal loadings, the beneficial influence of a ligand “double-layer” (coating of nanoparticles and the support by organic ligands) enhancing the adhesion between the Pt nanoparticles and the underlying monolithic support will be discussed. In the case of the metallic Ni substrate, this approach furthermore allows to circumvent alloy formation and nanoparticle diffusion into the metallic substrate. This can greatly increase long-term stability of systems coated directly onto metallic substrates without an additional oxidic washcoat.
Introduction
Whereas monolithic catalysts are already intensively used in automobile exhaust treatment1 and other combustion reactions (e.g. of fuel gas),2 industrial applications are still more rare. In view of their many advantages,1,3 however (e.g. lower energy input, high safety, easy catalyst separation) including lower pressure drops and improved mass transfer as compared to classical reactors filled with pellet beds,4 monolithic catalysts attract more and more attention in particular for low- or medium-temperature reactions, such as methanol steam reforming,5 oxidative dehydrogenation of propane,6 and liquid phase hydrogenations.7Although there are monolithic materials available which exhibit specific surface areas that are large enough for these types of applications8 (e.g. porous materials such as foams9 or aerogels10), more common materials, as also employed for exhaust catalysis, are low-surface area cordierite honeycombs,2,11 having the advantage of high thermal and mechanical stability. In order to increase their surface areas,5 such structures are often washcoated with oxides (e.g. with alumina). An alternative concept is the use of washcoated metallic monoliths12 exhibiting a superior thermal conductivity, which is often advantageous from a process engineering point of view (e.g.alumina-coated steel).13
Besides the need to synthesize tailored, well-defined monolithic structures with optimized mass and heat transfer properties and low pressure drops,14,15 the coating of the monolith with the catalytically active phase—often in the form of metal nanoparticles—must be optimized and improved.
Nowadays, traditional methods (e.g.dip-coating2 or impregnation16) are predominantly employed, i.e.nanoparticles of a catalytically active metal are grown onto the monolith or a pre-deposited washcoat by deposition of a suitable precursor1 and subsequent calcination and reduction steps. Another option is the incorporation of the precursors into the monolith material during its preparation17 or into a washcoat slurry.18 These approaches might, however, result in partial inclusion of the active phase. In any event, a homogeneous distribution of the active component on the monolith is often difficult to achieve.19,20
In addition to that, insufficient adhesion of the active material to the monoliths can be a problem leading to enhanced mechanical abrasion21 or leaching during catalytic reactions in solvents.22 Furthermore, after formation of the nanoparticles, sintering23 or aggregation24 can result in severe activity losses if the particles are not sufficiently anchored to the surface of the monolith.
In view of these shortcomings another approach which has just recently25 been described in literature seems promising: the coating of monoliths with preformed nanoparticles. Since colloidal nanoparticle synthesis allows for exceptional control of structural properties, such as size and shape of the nanoparticles26 (i.e., parameters that are known to strongly influence catalytic performance27,28), the use of preformed particles29,30 opens the way to the preparation of tailored catalytic systems. In the case of powder supports, the use of colloidal nanoparticles has already been shown to yield highly active catalysts.30–33
Against this background, the aim of this study is to present facile routes to coat different types of monoliths with colloidally synthesized nanoparticles with and without additional washcoats and to obtain highly active monolithic catalyst systems in this way.
First, since in the case of oxidic supports specific metal support interactions can sometimes play an important role for catalytic applications32,34 but the number of materials available in the form of oxidic monoliths is limited, we explored whether colloidally synthesized nanoparticles can also be easily codeposited onto monolithic structures together with a washcoat slurry. Specifically, we show that the high catalytic activity previously observed for colloidal NiPt particles on MgO powder32 can be transferred to a cordierite honeycomb by using a MgO washcoat.
In a second approach, we deal with the question whether such particles can be directly applied to monolithic supports. For this purpose, three monolithic supports were chosen—a classical cordierite honeycomb, an α-Al2O3 foam and a Ni fleece—all of which were coated with Pt nanoparticles without using a washcoat. Whereas the cordierite honeycomb is of interest due to its widespread applications in industry,2,11 the Al2O3 foam allows for a direct comparison with Pt/Al2O3 powder catalysts which have been characterized in detail throughout the literature.33,35,36 The third support material, the Ni fleece, was included as a (cheap) metallic substrate in order to explore options to coat metallic monoliths.
Whereas the NiPt nanoparticles were synthesized via a classical colloidal method,37 the Pt nanoparticles were obtained by the ethylene glycol method38 (for details see Experimental section), a colloidal synthesis route that yields nanoparticles without organic stabilizers, offering the option to modify the particles with a variety of organic ligands in a subsequent step.39 Using both types of particles enables us in the case of the ligand-free particles to directly compare with monolithic catalysts obtained by traditional methods and in the case of the ligand-covered particles to study the potential benefit of ligand-cappings while keeping the size and shape of the nanoparticles unaltered. In the current work, the latter aspect was studied in particular with respect to high metal loadings and the coating of metallic supports.
In the case of standard powder oxide supports, we could show in previous studies,32,33,40,41 that ligands such as dodecyl amine (DDA)—although decreasing the number40 and lowering the accessibility42 of active sites on the nanoparticles—can have an overall beneficial effect on heterogeneous gas phase catalysis. For instance, ligands were found to effectively prevent surface oxidation of the nanoparticles,40 enhance the anchoring to the support,43 and allow to tune strong metal–support interactions.41
Nanoparticles directly deposited onto a metallic monolith often suffer from alloy formation or nanoparticle diffusion into the substrate under reaction conditions.13,44 One way out of this dilemma is the use of metallic substrates with an oxidic washcoat45,46 as mentioned above. Nevertheless, achieving a good adhesion between the oxidic washcoat and the metallic monolith often causes difficulties since their thermal expansion coefficients usually differ strongly.3,47 Here, we present another strategy, namely depositing ligand-capped Pt nanoparticles on the nickel substrate with organic ligands serving as a buffer layer between Pt and Ni to avoid alloy formation and circumvent the diffusion of Pt into the metallic monolith.
Results and discussion
Fig. 1 shows exemplary micrographs of the colloidally synthesized Pt and NiPt nanoparticles as well as of the monolithic supports used in this study. The transmission electron microscopic (TEM) images in Fig. 1A and B depict nanoparticles from a colloidal solution of ligand-free and DDA-capped Pt nanoparticles dropped onto a TEM grid, respectively. They confirm the existence of quasi-spherical particles with an average size of about 2.2 (±0.3) nm in both cases. The NiPt nanoparticles used in this study (Fig. 1C) were also spherical in shape with an average size of 3.6 (±0.4) nm. In Fig. 1D–F overview scanning electron microscopic (SEM) images of the uncoated monolithic supports are depicted. Whereas the cordierite honeycomb (Fig. 1D) and the α-Al2O3 foam (Fig. 1E) were used as obtained, the Ni fleece (Fig. 1F) was coiled up prior to coating (Fig. 2). While the cordierite honeycomb (400 cpsi, Corning Inc.) and the Ni fleece were commercially available substrates, the alumina foam was obtained from an α-Al2O3 suspension viaalkane phase emulsification prior to foaming and sintering (for details see Experimental section).48 | ||
| Fig. 1 TEM images of colloidal solutions of (A) ligand-free Pt nanoparticles, (B) DDA-capped Pt nanoparticles, (C) NiPt nanoparticles capped with oleyl amine and oleic acid. SEM images of the uncoated monolithic (D) cordierite honeycomb, (E) α-Al2O3 foam and (F) of the Ni fleece. | ||
 | ||
| Fig. 2 Photograph of the monolithic supports used in this study. From left to right: cordierite honeycomb (400 cpsi), α-Al2O3 foam and coiled up Ni fleece. The weight of each monolith was adjusted to 1 g. | ||
All monolithic substrates used in this study had a specific surface area of roughly 1 m² g−1 according to BET measurements. The metal loading of all samples was checked before and after catalytic experiments by atomic absorption spectroscopy (AAS), some of which are summarized in Table 1. Whereas the metal loading after catalytic use was identical with the initial value for all samples with “low” loading (up to 0.025 wt%), the AAS measurements revealed for some samples with “high loading” (0.6 wt%) a significant loss of Pt.
| Monolith | Coating | Pt loading (wt%)a |
|---|---|---|
| a The initial Pt loading (as determined by means of AAS) of all samples was ∼0.6 wt%. b Both, the support and the Pt nanoparticles, were covered with dodecyl amine (DDA; for details see Experimental section). c The Pt nanoparticles were capped with DDA, while the Ni fleece was capped with lauric acid (LA; for details see Experimental section). | ||
| Cordierite honeycomb | ligand-free Pt | 0.50 |
| DDA–Pt | 0.41 | |
| DDA-“double-capped”b | 0.63 | |
| α-Al2O3 foam | ligand-free Pt | 0.29 |
| DDA–Pt | 0.49 | |
| DDA-“double-capped”b | 0.63 | |
| Nickel fleece | ligand-free Pt | 0.61 |
| DDA–Pt | 0.42 | |
| DDA-LA-“double-capped”c | 0.61 |
Catalytic activity of NiPt supported on cordierite honeycomb
As pointed out in the Introduction, specific metal–support interactions sometimes have a beneficial influence on the catalytic performance and can dramatically increase activity and/or change selectivities.34,41,49,50Thus, the challenge arises to transfer these beneficial catalytic properties from powders to monolithic structures. As the synthesis of monoliths from the required powders is not always possible, deposition of a slurry containing the powdered catalyst onto e.g. cordierite honeycombs is an alternative. Furthermore, such a washcoat has the advantage of a higher surface area as compared to the pristine honeycomb.
To this end, we present an approach to coat monolithic structures by codeposition of colloidally synthesized nanoparticles and an oxidic washcoat. Bimetallic NiPt nanoparticles obtained by a colloidal route37 were chosen, as some of us previously reported32 high CO oxidation activities of such particles—corroborated later on in this study by the alloying of the Pt nanoparticles with the Ni fleece. Noticeably however, in this former study distinct differences were observed for their catalytic activity in dependence of the underlying support.32 Briefly, due to the spillover of some of the amine ligands to a MgO support, the number of accessible active sites on the NiPt nanoparticles was increased whereas the remaining amine ligands on the NiPt surface were still able to prevent the Ni from being oxidized. Thus, when supported on MgO a much higher CO oxidation activity could be obtained as compared to γ-Al2O3.
Nevertheless, we started with direct deposition of NiPt nanoparticles onto a cordierite honeycomb (0.0025 wt%). To minimize the amount of the precious metal, a strongly diluted colloidal solution of ligand-capped NiPt nanoparticles was applied to the honeycomb structure and the solvent removed at room temperature. During deposition the honeycomb was rotated to obtain a homogeneous distribution. This easy approach led to an active monolithic catalyst.
As shown in Fig. 3, the activity of NiPt/cordierite was only slightly lower than reported for identical NiPt nanoparticles supported on γ-Al2O3 powder before.32 A turnover frequency (TOF) of ∼0.3 s−1 is reached with the cordierite supported sample at 180 °C, compared to 170 °C for the powder support. Thus, this approach allows for the preparation of monolithic catalysts with a similar activity compared to powder samples.
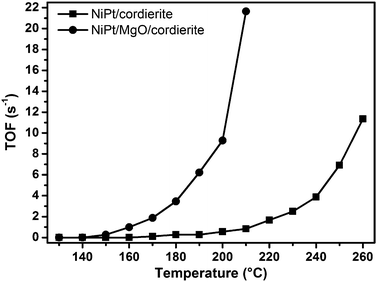 | ||
| Fig. 3 Turnover frequencies (TOFs) for the oxidation of CO on bimetallic NiPt nanoparticles (with oleyl amine and oleic acid as ligands) directly supported on a cordierite honeycomb and with an additional MgO washcoat. | ||
However, since MgO-supported NiPt—as already mentioned—was shown32 to have a much better performance than γ-Al2O3, in a further experiment a MgO washcoat was applied to the cordierite honeycomb, also to increase its surface area. Although the washcoating was performed by simple deposition of a slurry containing MgO and a colloidal NiPt solution onto the honeycomb (see Experimental section), the activity of the obtained monolithic catalyst indeed strongly improved compared to the sample without washcoat (Fig. 3). The TOF reported for a sample supported on MgO powder (14.6 s−1 at 170 °C), however, could not be reached with this approach. Probably, due to the washcoating procedure, part of the NiPt nanoparticles was embedded in the MgO and thus not accessible for catalytic reactions thus lowering the number of active surface sites and accordingly the TOFs. Possibly, a more elaborate form of deposition or a sequential deposition of the MgO slurry and the nanoparticles would lead to a further increase of catalytic activity.
Catalytic activity of Pt supported on cordierite honeycomb
After demonstrating that highly active monolithic catalysts can be obtained in the special case of colloidal NiPt nanoparticles directly deposited onto cordierite honeycombs, we studied in a second series of experiments whether sufficiently active systems can also be obtained by depositing intrinsically less active nanoparticles without washcoat onto low surface area monolithic supports. As a counterpart to NiPt, we prepared ligand-free and DDA-capped pure Pt nanoparticles and deposited them onto the cordierite honeycomb. Different initial Pt loadings from 0.025 wt% to 0.6 wt% were realized by dipping the cordierite honeycombs into the colloidal solution and evaporating the solvent completely (for details see Experimental section).The CO oxidation activities of the samples coated with ligand-free Pt nanoparticles (lowest and highest loading, respectively) are displayed in Fig. 4 as square symbols. Whereas in the case of low Pt loadings the amount of converted CO was stable at a given temperature, the activity of the catalyst with the highest Pt loading of 0.6 wt% varied during the first catalytic run indicating structural changes. Therefore, the stable activities in the second and subsequent runs are exemplary plotted in Fig. 4.
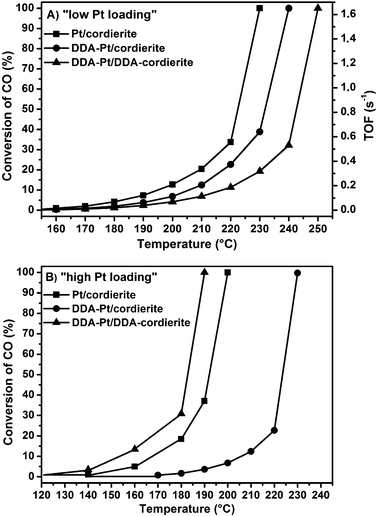 | ||
| Fig. 4 CO oxidation activity of different ligand-free and ligand-capped samples supported on monolithic cordierite honeycombs. Initial Pt loading of (A) 0.025 wt% and (B) 0.6 wt% (as determined by AAS). | ||
Clearly, the activity can be significantly increased as the loading is increased finally allowing even similar conversions as in the case of powder samples. (Note that 0.6 wt% is already in the regime of typical loadings for high-surface area powders.) TEM images (Fig. 5D) reveal however that some agglomeration takes place leading to large islands of Pt on the cordierite support. Another more severe problem associated with such high coverages is revealed by the AAS results summarized in Table 1. In contrast to the samples with lower metal loadings, the Pt content after catalysis is apparently noticeably reduced for the sample with an initial loading of 0.6 wt%. Although the formation of volatile compounds of Pt, such as PtO2, under the conditions applied in the catalytic reactor cannot be fully excluded, a part of the nanoparticles is likely to have been carried away by the flowing gas (50 mL min−1) due to weak adhesion at these high coverages of the low-surface area monoliths.51,52
 | ||
| Fig. 5 TEM images of Pt nanoparticles capped with DDA (A–C) and ligand-free (D) deposited on low surface area cordierite honeycomb. Metal loading: (A) 0.025 wt%, (B) 0.3 wt% (C) and (D) 0.6 wt% (Please note that areas without particles (such as in B) represent fracture surfaces of the cordierite that were not accessible to the nanoparticles during deposition.) | ||
For this reason, another approach was taken by using ligand-capped nanoparticles. On the one hand, it is known that ligands such as DDA can influence catalytic activity (and selectivity) covering the nanoparticles or the support.40–42 On the other hand, a beneficial effect with respect to adhesion and protection against sintering can be expected. To disentangle these effects, different situations were studied: ligand-capped nanoparticles were deposited on the pristine cordierite as well as on a sample precapped with the organic ligand (“double-capped” situation). Different metal loadings between 0.025 wt% and 0.6 wt% were again realized. TEM images (Fig. 5A–C) reveal that only in the case of the highest loading some agglomeration of the particles occurs, being, however, significantly weaker than in the case of the ligand-free particles—a fact that clearly points to a protecting function of the ligands against agglomeration (compare with Fig. 5D; note the different scale bar as compared to 5C). In contrast, up to a loading of about 0.3 wt% the size of the DDA-capped Pt nanoparticles remains in the range of 2–3 nm although the particle density is already quite high (see Fig. 5B).
Turning to the samples with low metal loadings first (Fig. 4A), the ligand-free Pt has the highest activity whereas the “double-capped” sample is the least active. This trend—in agreement with results obtained for colloidal Pt nanoparticles on high-surface area powder supports33,40—can of course be straightforwardly explained by an increasing amount of ligands lowering the number of active Pt sites available for the catalytic reaction.40,42 The obtained TOF values are in the same range as those obtained for Pt single crystals and colloidally synthesized Pt nanoparticles under similar conditions.33,53
For the highest Pt loading (0.6 wt%) the trend changes, though (Fig. 4B). In this case, the activity decreases in the following order: “double-capped” > ligand-free ≫ “single-capped”. The observed trend is reflected by the AAS data after catalysis, showing that the differences are caused by different losses of Pt during the first catalytic cycle: no loss of Pt was found in the case of the “double-capped” samples, whereas the loss was highest for the ligand-capped particles deposited on the pristine cordierite followed by the ligand-free particles.
The results thus prove that, when coatings with very high particle densities are realized on low-surface area monoliths, the different adhesion properties of nanoparticles with and without ligand shell come into play. While in the case of low loadings defects and pores provide enough anchor sites to hold ligand-free and ligand-capped particles, this is not the case for high loadings. Here, the surface chemistry of the monolith material becomes important. The surface of cordierite (2MgO·Al2O3·5SiO2) is known to be acidic, since Si strengthens the Lewis acidic properties of Al while simultaneously compensating the basicity of Mg.54 It is understandable that the adhesion of the ligand-capped Pt on the pristine cordierite surface is the weakest, as it is not likely that the cordierite surface (dominated by Lewis and Brønsted acidic sites) interacts significantly with the lipophilic ligand shell. On the other hand, as exposure to moisture can lead to the transformation of some Lewis acidic sites into Brønsted acidic surface hydroxyl groups that are known to interact strongly with small Pt particles,55 an intermediate adhesion is expected for the ligand-free Pt nanoparticles as indeed observed.
For the “double-capped” sample, the good adhesion (no Pt loss during the catalytic runs) can be related to the interaction of the nitrogen lone pair of the amine with Lewis acidic Al sites on the cordierite. DDA was found to have a strong tendency to bind to Al2O3.40 In turn, the surface of the monolith becomes lipophilic because of the hydrocarbon chain of DDA and a “lipophilic double layer” providing optimized adhesion can form with the ligand-shell of the capped Pt nanoparticles.
Catalytic activity of Pt particles supported on α-Al2O3 foam
In order to check whether such an optimization of adhesion properties by functionalization of nanoparticles and the support, is also applicable to realize high metal loadings on other low-surface area monolithic systems, a second series of Pt samples was prepared and deposited on a monolithic Al2O3-foam obtained from α-Al2O3 powder via an alkane phase emulsion by direct foaming as described by Barg et al.48 Again, three samples (ligand-free, ligand-capped and “double-capped”) with an initial Pt loading of 0.6 wt% were synthesized by complete evaporation of the colloidal solution (for details see Experimental section).All three monoliths were catalytically studied with respect to CO oxidation; the resulting conversions are displayed in Fig. 6 as a function of temperature. Again, the activity of the second catalytic cycles are displayed, since the catalytic activity of the samples was not constant at a given temperature during the first heating cycle—apparently again due to structural changes of the pristine samples under reaction conditions.
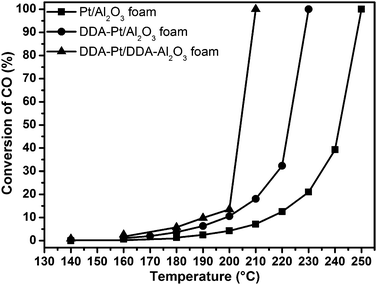 | ||
| Fig. 6 CO oxidation activity of different ligand-free and ligand-capped samples supported on monolithic α-Al2O3 foams. | ||
Like in the case of the cordierite supported Pt, the “double-capped” sample is the most active one although the order of the other two samples is changed for the Al2O3 support as compared to the cordierite honeycomb. However, again the results can be correlated with the observed Pt loss following the same trend than the activities (compare with Table 1). Thus, the catalytic activity of the Pt samples supported on the monolithic α-Al2O3 foams decreases with decreasing Pt amount. As in the case of the cordierite honeycombs, only the metal loading of the “double-capped” sample did not change during the measurements and maintained its initial value of 0.63 wt%.
Again, the observed trends can be correlated with the surface properties of the α-Al2O3 monolith. Optimized adhesion in the case of the “double-capped” sample can be explained in the same way as for the cordierite: as already reported in a previous study40 the amine ligand binds strongly to Lewis acidic Al sites via the lone pair of the nitrogen atom so that the lipophilic hydrocarbon chain pointing away from the surface can interact with the lipophilic ligand-shell of the capped Pt nanoparticles.
The intermediate adhesion of the amine-capped Pt on the pristine α-Al2O3 foam (with a lower loss of Pt as compared to the same particles on cordierite) might be understood in terms of the relatively low density of hydroxyl groups on the α-Al2O3 surface providing a higher hydrophobicity than cordierite.56
The weak binding of the ligand-free Pt to the surface of the monolithic α-Al2O3 foam is reasonable since it is mainly terminated by electron-deficient Lewis acid Al sites which do not tend to interact strongly with (the slightly positively charged) surface of the Pt nanoparticles. In turn, in contrast to the cordierite the lower number of (Brønsted acidic) hydroxyl groups does apparently not lead to a sufficient adhesion of ligand-free Pt nanoparticles in the case of high metal loadings.
Catalytic activity of Pt supported on Ni fleece
Turning to the nickel fleece, again three samples (ligand-free, ligand-capped and “double-capped”) were prepared with the aim to compare alloy formation, diffusion and sticking behaviour. However, the coating of the Ni fleece in the case of the “double-capped” sample was realized with lauric acid instead of DDA because Ni is known to preferably bind carboxylic acids. (Please note that the two ligands possess identical hydrocarbon chains and differ only in their functional groups.)The pristine ligand-free Pt on the Ni fleece exhibited by far the highest activity (full conversion already at 140 °C) of all investigated samples (Fig. 7A). This can be related to NiPt alloy formation44 leading to the same bimetallic effect as in the case of NiPt nanoparticles and thus enhanced catalytic activity as compared to monometallic Pt.32 However, after 8 h on stream the activity severely decreased and higher temperatures were needed to maintain full conversion of CO for some more hours before a decrease was again observed. Most likely, this behaviour can be attributed to the diffusion of Pt into the Ni bulk at elevated temperatures and over a prolonged period of time. However, in contrast to the recently reported57 segregation of Pt into the core of bimetallic NiPt nanoparticles, this diffusion process could not be reversed by H2 treatment (data not shown).
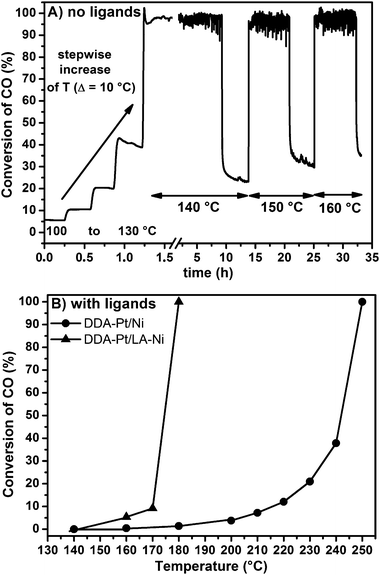 | ||
| Fig. 7 CO oxidation activity of (A) a pristine ligand-free Pt coated Ni fleece and (B) DDA-capped Pt on Ni fleece and a corresponding “double-capped” sample. | ||
Turning to the ligand-capped samples (Fig. 7B) supported on Ni fleece, the “double-capped” sample had a superior activity compared to the “single-capped” DDA–Pt/Ni fleece. AAS measurements (Table 1) confirmed that in the case of the “single-capped” sample some Pt was lost during the catalytic runs whereas both for the “double-capped” as well as for the ligand-free samples the metal loading did not decrease. Whereas the good adhesion of the former sample due to the ligand bilayer (lauric acid binds to Ni, DDA to Pt) needs no further explanation, the unchanged Pt content in the latter case proves that indeed Pt diffused into the Ni fleece and did not get lost via other routes. The weak adhesion in the case of the “single-capped” sample can again be ascribed to the lipophilic nature of the ligands not attracted by metallic surfaces.
For better comparison, a pristine ligand-free Pt/Ni fleece was heated to 180 °C under CO oxidation conditions. Full conversion was immediately reached, but after ∼5 h a sharp decrease to only 40% conversion occurred. In the next 12 h the activity decreased slowly over time to only 20%. In contrast, a pristine “double-capped” sample heated to 180 °C maintained its high activity (full conversion of CO) for at least four weeks. This proves that alloy formation and diffusion can be effectively prevented by the ligand bilayer even at relatively high temperatures and under reaction conditions under which ligand decomposition can be expected to play a role (please note that TGA measurements revealed that DDA starts to decompose/desorb at temperatures of roughly 200 °C).40
Additionally, we performed experiments with oleylamine and oleic acid instead of DDA and lauric acid as these ligands have also been used for the colloidal synthesis of the bimetallic NiPt nanoparticles. The obtained results (data not shown for brevity) were almost identical to those shown in Fig. 7B although—most likely due to the double bond of the unsaturated ligands being more vulnerable to O2 at higher temperatures—in the first run large amounts of CO2 were formed pointing to severe ligand decomposition (possibly leading to some carbonaceous species). However, the different ligands apparently do not change the overall catalytic behaviour. Apparently, on the one hand such a carbon-based layer effectively protects the nanoparticles against diffusion into the nickel fleece but on the other hand does not enclose them, so that the high initial activity of the Pt nanoparticles is conserved.
Conclusion
It was shown that highly active catalytic coatings on different types of monolithic structures can be obtained using colloidally prepared nanoparticles. Additionally, high catalytic activities that rely on specific metal–support interactions can be transferred from powder catalysts to monolithic structures by applying a facile coating procedure introduced in this study, namely the codeposition of colloidally synthesized nanoparticles and an oxidic washcoat slurry.Whereas the obtained results are comparable with those of powder catalysts in the case of low Pt loadings, agglomeration and adhesion of the nanoparticles to the monoliths were found to play crucial roles applying very high metal loadings on low surface area monoliths.
However, agglomeration can be significantly diminished and the adhesion enhanced by organic ligands applied to the particles and the support. This strategy leads to minimized agglomeration and optimized adhesion without severely blocking catalytically active sites on the surface of the nanoparticles so that high catalytic activity could be achieved. Furthermore, such a high amount of ligands (capping both nanoparticles and the monolith) might be beneficial in order to steer catalytic selectivity for certain reactions as was suggested in a recent study.58
In addition, our study reveals that organic ligands can serve as a buffer layer between Pt nanoparticles and a metallic support. This result is promising as it represents an approach to overcome particle diffusion into the metallic substrate and alloy formation as was observed when classical impregnation methods were applied. This concept, exemplified here for Pt-coated Ni fleece, should of course be applicable to other metallic substrates of high industrial relevance (e.g. steel) as well.
Interestingly, the decomposition of ligands over time does not seem to change the adhesion between the nanoparticles and the monolith or the function of a buffer layer in the case of the metallic substrate. Thus, most likely this approach is also suitable for applications under which the ligand shell is not longer in its initial form (like e.g. intact DDA molecules). In future studies, however, the nature of the remaining—probably carbonaceous—buffer layer has to be investigated in detail.
Experimental section
The colloidal synthesis of NiPt nanoparticles stabilized with a mixture of oleyl amine and oleic acid is described in detail elsewhere.37Pt nanoparticles were prepared in a way similar to one reported previously.38 In a typical experiment, an ethylene glycol (EG) solution of H2PtCl6·6H2O (50 mL, 41 mM) was added into an EG solution of NaOH (50 mL, 400 mM) under stirring to obtain a transparent yellow platinum hydroxide or oxide colloidal solution, which was then heated at 160 °C for 3 h. A transparent dark-brown homogeneous colloidal solution of Pt metal nanoparticles (20.5 mM, 4.0 g Pt per L) was obtained without any precipitation.
Prior to the deposition of ligand-free Pt nanoparticles onto the monolithic supports, the nanoparticles were precipitated from the EG solution by adding an aqueous solution of 1 M HCl. After centrifugation, the obtained precipitates of ligand-free Pt nanoparticles were washed with the 1 M HCl three times and then redispersed in acetone.
To obtain Pt nanoparticles capped with dodecylamine (DDA) a method described by Fu et al.39 was adapted. A diluted EG solution (10 mL) of ligand-free Pt nanoparticles (10.3 mM) was added dropwise into a toluene solution (10 mL) of 20.5 mM DDA. The obtained mixture of two phases was stirred at room temperature for 3 h and then rested for 1 h to form distinct two phases. The EG phase was removed and the toluene phase was washed with water three times, which was then concentrated to ∼0.5 mL under flowing N2. To this concentrated solution, 10 mL of ethanol were added to precipitate the products, which were subsequently isolated by centrifugation and washed with ethanol three times to remove free ligands. The washed precipitates of DDA-capped Pt nanoparticles were finally dissolved in chloroform.
Whereas the cordierite (400 cpsi, Corning Inc.) and the Ni fleece were commercially available, the α-Al2O3 foam was produced via an alkane phase emulsion in a direct foaming process described in detail elsewhere.48
Prior to deposition of the nanoparticles, some of the monoliths were capped with ligands by putting the monoliths into a toluene solution of the respective ligand and saturating it for 5 h under ultrasonification.
The deposition of Pt nanoparticles onto the monoliths was performed by adding the monolith to a colloidal solution under ultrasonification until solvent evaporation was complete. The monoliths were rotated regularly to ensure homogeneous coating. To minimize the amount of metal (only 17.6 μg per g (cordierite) in the case of NiPt/MgO/cordierite) the monoliths were coated with NiPt nanoparticles in a slightly different way by injecting dilute colloidal solution or a NiPt/washcoat slurry with a syringe, respectively.
Transmission electron microscopy (TEM) measurements were carried out by using a Tecnai F20 S-TWIN microscope (FEI) operated at 200 kV. Samples for TEM measurements were prepared by placing a drop of the diluted EG solution of Pt nanoparticles on a carbon-coated copper grid. Scanning electron microscopic measurements were performed with a Supra 40 (Zeiss) after coating with Au for 15 s. Atomic absorption spectroscopy (AAS) measurements were conducted on finely ground powder obtained from a part of the monolithic catalysts to analyze the metal loading before and after catalytic measurements. For AAS measurements an AAS 5FL Flame from Zeiss was used and operated with an acetylene/nitrous oxide flame after dissolving the samples in aqua regia, boiling for 1 min and leaving them overnight.
Catalytic measurements were carried out with monolithic samples (∼1 g) with quartz wool below and on top. When possible, the diameter of the samples was chosen to be as close to the inner diameter of the quartz tube used as the catalytic reactor as possible. This way, the gas by-pass was minimized as can also be deducted from the full CO conversion of the samples. For CO oxidation, a gas flow of 50 mL min−1 containing 3 vol% of CO in synthetic air was used. The components in the product stream were analyzed with a photometric detector for CO/CO2 (URAS 3G, Hartmann & Braun).
Turnover frequencies (TOFs) were calculated by assuming a spherical particle shape and accessibility of all nanoparticle surface atoms. Because all particles are in contact with the support and a part of their surface is not available for CO oxidation, the calculated TOF values represent lower limits.
Acknowledgements
We thank Dr Oliver Seel (BASF) for supplying us with the nickel fleece, Dr-Ing. Jens Hüppmeier from the group of Prof. Dr-Ing. Jörg Thöming (Center for Environmental Research and Sustainable Technology—UFT) in Bremen for providing the alumina foam and PD Dr Naoufal Bahlawane for the supply of the cordierite honeycomb. Furthermore we appreciate the help of Dr Kasten Thiel, PD Dr Volkmar Zielasek and Willian G. Menezes (Fraunhofer Institute for Manufacturing Technology and Applied Materials Research (IFAM) in Bremen) and Petra Witte (Research Group Historical Geology—Palaeontology, University of Bremen) for performing the TEM and SEM measurements, respectively.References
- T. A. Nijhuis, A. E. W. Beers, T. Vergunst, I. Hoek, F. Kapteijn and J. A. Moulijn, Catal. Rev. Sci. Eng., 2001, 43, 345–380 CrossRef CAS.
- J. W. Geus and J. C. van Giezen, Catal. Today, 1999, 47, 169–180 Search PubMed.
- P. Avila, M. Montes and E. E. Miró, Chem.–Eng. J., 2005, 109, 11–36 CrossRef CAS.
- F. C. Patcas, G. Incera Garrido and B. Kraushaar-Czarnetzki, Chem. Eng. Sci., 2007, 62, 3984–3990 Search PubMed.
- B. Lindström and L. J. Pettersson, J. Power Sources, 2002, 106, 264–273 CrossRef CAS.
- G. Capannelli, E. Carosini, F. Cavani, O. Monticelli and F. Trifiro, Chem. Eng. Sci., 1996, 51, 1817–1826 Search PubMed.
- T. Boger, M. M. P. Zieverink, M. T. Kreutzer, F. Kapteijn, J. A. Moulijn and W. P. Addiego, Ind. Eng. Chem. Res., 2004, 43, 2337–2344 CrossRef CAS.
- P. C. Stair, J. Chem. Phys., 2008, 128, 182507 Search PubMed.
- J. Luyten, S. Mullens, J. Cooymans, A. M. De Wilde, I. Thijs and R. Kemps, J. Eur. Ceram. Soc., 2009, 29, 829–832 Search PubMed.
- Y. Tokudome, K. Nakanishi, K. Kanamori, K. Fujita, H. Akamatsu and T. Hanada, J. Colloid Interface Sci., 2009, 338, 506–513 CrossRef CAS.
- R. M. Heck, S. Gulati and R. J. Farrauto, Chem. Eng. J., 2001, 82, 149–156 CrossRef CAS.
- M. Liu, W. Yu, H. Liu and J. Zheng, J. Colloid Interface Sci., 1999, 214, 231–237 CrossRef CAS.
- A. S. Pratt and J. A. Cairns, Platinum Met. Rev., 1977, 21, 74–83 Search PubMed.
- P. Colombo, E. Bernardo and L. Biasetto, J. Am. Ceram. Soc., 2004, 87, 152–154 Search PubMed.
- M. K. Schröter, L. Khodeir, J. Hambrock, E. Löffler, M. Muhler and R. A. Fischer, Langmuir, 2004, 20, 9453–9455 CrossRef CAS.
- A. A. Klinghoffer, R. L. Cerro and M. A. Abraham, Catal. Today, 1998, 40, 59–71 Search PubMed.
- L. Y. Lee, S. P. Perera, B. D. Crittenden and S. T. Kolaczkowski, Adsorpt. Sci. Technol., 2000, 18, 147–170 Search PubMed.
- R. Ran, G. Xiong and W. Yang, J. Mater. Chem., 2002, 12, 1854–1859 RSC.
- A. C. S. Samia, J. A. Schlueter, J. S. Jiang, S. D. Bader, C.-J. Qin and X.-M. Lin, Chem. Mater., 2006, 18, 5203–5212 CrossRef.
- A. Wahlberg, L. J. Pettersson, K. Bruce, M. Andersson and K. Jansson, Appl. Catal., B, 1999, 23, 271–281 Search PubMed.
- H. Wichmann, G. A. K. Anquandah, C. Schmidt, D. Zachmann and M. A. Bahadir, Sci. Total Environ., 2007, 388, 121–127 CrossRef CAS.
- C.-S. Chen, H.-W. Chen and W.-H. Cheng, Appl. Catal., A, 2003, 248, 117–128 CrossRef CAS.
- W. Ruettinger, X. Liu and R. J. Farrauto, Appl. Catal., B, 2006, 65, 135–141 CrossRef CAS.
- K.-I. Tanaka, M. Shou, H. He and X. Shi, Catal. Lett., 2006, 110, 185–190 Search PubMed.
- D. Connolly, B. Twamley and B. Paull, Chem. Commun., 2010, 46, 2109–2111 RSC.
- N. Semagina and L. Kiwi-Minsker, Catal. Rev.-Sci. Eng., 2009, 51, 147–217 CrossRef CAS.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097–3101 CrossRef CAS.
- M. Grass, R. Rioux and G. Somorjai, Catal. Lett., 2008, 128, 1–8 Search PubMed.
- A. Berenguer, M. Cantoro, V. B. Golovko, S. Hofmann, C. T. Wirth, B. F. G. Johnson and J. Robertson, Phys. Status Solidi B, 2009, 246, 2436–2439 Search PubMed.
- S. Domínguez-Domínguez, Á. Berenguer-Murcia, D. Cazorla-Amorós and Á. Linares-Solano, J. Catal., 2006, 243, 74–81 CrossRef CAS.
- M. Comotti, W.-C. Li, B. Spliethoff and F. Schüth, J. Am. Chem. Soc., 2006, 128, 917–924 CrossRef CAS.
- B. Jürgens, H. Borchert, K. Ahrenstorf, P. Sonström, A. Pretorius, M. Schowalter, K. Gries, V. Zielasek, A. Rosenauer, H. Weller and M. Bäumer, Angew. Chem., Int. Ed., 2008, 47, 8946–8949 CrossRef.
- D. Fenske, P. Sonström, J. Stöver, X. Wang, H. Borchert, J. Parisi, J. Kolny-Olesiak, M. Bäumer and K. Al-Shamery, ChemCatChem, 2010, 2, 198–205 Search PubMed.
- M. Haruta and M. Daté, Appl. Catal., A, 2001, 222, 427–437 CrossRef CAS.
- R. H. Nibbelke, M. A. J. Campman, J. H. B. J. Hoebink and G. B. Marin, J. Catal., 1997, 171, 358–373 CrossRef CAS.
- A. Bourane and D. Bianchi, J. Catal., 2003, 218, 447–452 Search PubMed.
- K. Ahrenstorf, O. Albrecht, H. Heller, A. Kornowski, D. Görlitz and H. Weller, Small, 2007, 3, 271–274 CrossRef CAS.
- Y. Wang, J. Ren, K. Deng, L. Gui and Y. Tang, Chem. Mater., 2000, 12, 1622–1627 CrossRef CAS.
- X. Fu, Y. Wang, N. Wu, L. Gui and Y. Tang, J. Colloid Interface Sci., 2001, 243, 326–330 CrossRef CAS.
- X. Wang, P. Sonström, D. Arndt, J. Stöver, V. Zielasek, H. Borchert, K. Thiel, K. Al-Shamery and M. Bäumer, J. Catal., 2011, 278, 143–152 Search PubMed.
- P. Sonström, D. Arndt, X. Wang, V. Zielasek and M. Bäumer, Angew. Chem., Int. Ed., 2011, 50, 3888–3891 Search PubMed.
- A. Quintanilla, V. C. L. Butselaar-Orthlieb, C. Kwakernaak, W. G. Sloof, M. T. Kreutzer and F. Kapteijn, J. Catal., 2010, 271, 104–114 CrossRef CAS.
- P. Sonström, M. Adam, X. Wang, M. Wilhelm, G. Grathwohl and M. Bäumer, J. Phys. Chem. C, 2010, 114, 14224–14232 Search PubMed.
- D. A. MacLaren and W. Allison, J. Phys.: Condens. Matter, 2005, 17, 7455–7463 Search PubMed.
- J. F. Sánchez M, O. J. González Bello, M. Montes, G. M. Tonetto and D. E. Damiani, Catal. Commun., 2009, 10, 1446–1449 Search PubMed.
- B. Kucharczyk and W. Tylus, Catal. Lett., 2007, 115, 122–132 Search PubMed.
- H. Sun, X. Quan, S. Chen, H. Zhao and Y. Zhao, Appl. Surf. Sci., 2007, 253, 3303–3310 Search PubMed.
- S. Barg, C. Soltmann, M. Andrade, D. Koch and G. Grathwohl, J. Am. Ceram. Soc., 2008, 91, 2823–2829 Search PubMed.
- A. Dandekar and M. A. Vannice, J. Catal., 1999, 183, 344–354 CrossRef CAS.
- Y. N. Sun, Z. H. Qin, M. Lewandowski, E. Carrasco, M. Sterrer, S. Shaikhutdinov and H. J. Freund, J. Catal., 2009, 266, 359–368 CrossRef CAS.
- P. M. J. A. Hermans, Z. I. Kolar, J. J. M. De Goeij, F. Kapteijn and J. A. Moulijn, Appl. Radiat. Isot., 1997, 48, 1521–1524 Search PubMed.
- J. Sungchul and F. B. Daniel, Nanotechnology, 2010, 21, 055204 Search PubMed.
- A. K. Santra and D. W. Goodman, Electrochim. Acta, 2002, 47, 3595–3609 CrossRef CAS.
- M. I. Baraton, T. Merle-Mejean, P. Quintard and V. Lorenzelli, J. Phys. Chem., 1990, 94, 5930–5934 CrossRef CAS.
- Z. C. Zhang and B. C. Beard, Appl. Catal., A, 1999, 188, 229–240 CrossRef.
- S. Koonaphapdeelert and K. Li, J. Membr. Sci., 2007, 291, 70–76 Search PubMed.
- R. Mu, Q. Fu, H. Liu, D. Tan, R. Zhai and X. Bao, Appl. Surf. Sci., 2009, 255, 7296–7301 CrossRef CAS.
- S. T. Marshall, M. O'Brien, B. Oetter, A. Corpuz, R. M. Richards, D. K. Schwartz and J. W. Medlin, Nat. Mater., 2010, 9, 853–858 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |