ZnO based nanowires grown by chemical vapour deposition for selective hydrogenation of acetylene alcohols
L. N.
Protasova
a,
E. V.
Rebrov†
a,
K. L.
Choy
b,
S. Y.
Pung
b,
V.
Engels
c,
M.
Cabaj
c,
A. E. H.
Wheatley
c and
J. C.
Schouten
*a
aDepartment of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: j.c.schouten@tue.nl
bFaculty of Engineering, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
cDepartment of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK
First published on 26th May 2011
Abstract
Vertically aligned ZnO nanowires (NWs) with a length of 1.5–10 μm and a mean diameter of ca. 150 nm were grown by chemical vapour deposition onto a c-oriented ZnO seed layer which was deposited by atomic layer deposition on Si substrates. The substrates were then spin-coated with an ethanol solution containing Pd nanoparticles with an average size of 2.7 and 4.5 nm. A homogeneous distribution of the Pd nanoparticles on ZnO NWs has been obtained using both Pd particle series. The catalytic activity of the ZnO NWs and Pd/ZnO NWs catalysts was measured in the semihydrogenation of 2-methyl-3-butyn-2-ol at 303-343 K and a pressure of 2–10 bar. The effect of the solvent used on the catalytic performance of the Pd/ZnO NWs catalyst was studied. The Pd/ZnO catalysts showed alkene selectivity of up to 95% at an alkyne conversion of 99%. A kinetic model is proposed to explain the activity and selectivity of the ZnO support and Pd/ZnO catalysts.
1. Introduction
A variety of deposition techniques have been used to grow ZnO thin films, for example atomic layer deposition (ALD), molecular beam epitaxy, pulsed layer deposition, sol–gel method etc.1,2 The advantage of the ALD technique is the capability to control the crystal growth direction of highly textured ZnO thin films over metal nanoparticles deposited onto Si substrate.2ALD is based on the sequential pulsing of a chemical vapour precursor (from gas, liquid or solid source) into a mildly heated reactor where self-terminating gas–solid reactions take place and each pulse forms about one atomic layer. The growth of oxide nanostructures occurred either from the vapor phase at 1173 K3 or via a surface terminated chemical reaction at ca. 373 K.4 Recently, a new structured catalyst based on sintered metal fibers with high mechanical strength, high permeability, and low pressure drop properties has been developed for the selective hydrogenation of functionalized alkynes.5 This has proved to be an important step in the synthesis of fine chemicals,5–7 intermediates in the synthesis of vitamins A and E8 and terpenes.9 The support was coated by a grain-structured ZnO layer and loaded using a Pd0-sol of 7 nm nanoparticles. The new catalyst showed high activity, selectivity, and stability. During the mentioned reduction in hydrogen at 773 K, 24 nm crystallites of Pd0/PdZn alloy were formed. It was found that the Pd0/PdZn/ZnO phase was responsible for the catalyst activity, selectivity, and stability of the catalyst during the hydrogenation.5There has also been increased interest in developing novel synthesis approaches such as coating of nanostructured objects with nanoparticles,6 and the use of shape-selective carbon nanotubes as catalyst supports.10Zinc oxide nanostructures have been considered for several applications in catalysis, such as the synthesis of methanol, and hydrogenation of unsaturated hydrocarbons.1 The nanowire structure demonstrates a clear advantage over mesoporous films deposited on the inner channel walls of microstructured reactors, as they have much larger open porosity and an easily accessible structure.1,3 The latter increases considerably the effective diffusivity of large organic molecules into the porous domain, which in turn allows the increase of catalyst loadings far beyond the level of 3 kg m−3 that is typically associated with supported thin film catalysts in microchannels.11
The hydrogenation of 2-methyl-3-butyn-2-ol (MBY) to 2-methyl-3-buten-2-ol (MBE) is often used as a test reaction (Fig. 1). In industry, this reaction is performed in slurry reactors over a 5% Pd/CaCO3 powder catalyst modified by lead acetate, known as the Lindlar catalyst. A yield of ca. 97% of the semi-hydrogenated product can be achieved, however the catalyst deactivates after just a single cycle if water is present in the solvent. The Lindlar catalyst also suffers from the additional drawback that it must be separated from the reaction products. Using lead further limits its application in several industrial processes due to increasing safety restrictions. In seeking to overcome these limitations, experiments have been done in which selectivities of 95–97% towards alkenes in liquid-phase hydrogenation using supported Pd catalysts were obtained.5,6 For MBY hydrogenation in ethanol, the selectivity to MBE has been found to increase in the following order: Pd black < Pd/C < Pd/Al2O3 < Pd/BaSO4 < Pd/MgO < Pd/ZnO ≈ Pd/CaCO3.5 Two effects in Pd/ZnO have been proposed to be responsible for the improved selectivity: firstly, the metal-support interaction in case of reducible supports and, secondly, the electron donating effect from Zn, which is electropositive relative to Pd. The resulting increase in electron density of the noble metal enhances the selectivity due to decreased alkene adsorption.5,12 Selectivity to the alkene can also be improved by employing electron donor compounds, like N-bases (quinoline, pyridine, ammonia), which, however, increases separation costs in downstream processing. Pd and Pt supported nanostructured ZnO catalysts have demonstrated high selectivities towards the desired products in a number of catalytic reactions such as ester hydrogenation (99%),13hydrogenation of CO2 to methanol (99%),14 and the selective hydrogenation of cinnamaldehyde (96%)12 and crotonaldehyde (80%).15
 | ||
| Fig. 1 Reaction scheme of 2-methyl-3-butyn-2-ol hydrogenation and side reactions. MBY = 2-methyl-3-butyne-2-ol, MBE = 2-methyl-3-butene-2-ol, MBA = 2-methyl-3-butane-2-ol, 1 = 2,7-dimethyl-3,5-octadiyne-2,7-diol, 2 = 2,7-dimethyl-5-octen-3-yne-2,7-diol, 3 = prenal. | ||
Water, the universal solvent, has been recognized as one of the safest and most environmentally friendly alternatives to organic solvents.16 However, many nanostructures are not stable in water even under ambient conditions. For the past few years, several attempts have been made to synthesize water-stable ZnO nanoparticles.17,18
Here, we report a novel synthesis method to prepare Pd-based ZnO catalysts to selectively hydrogenate acetylene alcohols, based on vertically aligned ZnO nanowires grown by chemical vapour deposition (CVD) onto a c-oriented ZnO seed layer deposited by ALD which offers precise control of the thin film deposition down to the atomic scale.19 To the best of our knowledge, such CVD grown ZnO based nanowires whose activity is enhanced with Pd nanoparticles have not yet been used as heterogeneous catalysts. In this paper, two types of catalysts prepared by CVD based techniques (i.e.CVD and ALD) are compared: an unpromoted ZnO NWs catalyst and two Pd/ZnO NWs catalysts, nominally the same catalysts promoted with monodispersed Pd nanoparticles with an average size of 2.7 and 4.5 nm. The abovementioned catalysts were tested in MBY hydrogenation. The stability of the nanostructured catalysts was studied in methanol, water and a mixture thereof.
2. Experimental
2.1 Fabrication of Pd nanoparticles
0.23 g (1 mmol) Pd(OAc)2 was dissolved in 10 ml of 1,4-dioxane and the orange solution stirred at room temperature (RT). 0.81 g poly(N-vinylpyrrolidone) (PVP, M(average) = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; PVP/Pd ratio ca. 10:1) was dissolved in 120 ml of anhydrous ethylene glycol and the resulting solution was heated. After adjusting the pH to 9–11 (ca. 5 ml of 1 M NaOH), the palladium acetate solution was added and the reaction was stirred to yield a black colloidal suspension (batch 1, 100 °C for 1 h; batch 2, 70 °C for 2 h). Aliquots of the product were purified by extracting 50 ml suspensions from ethylene glycol and surplus polymer using excess acetone. After sedimentation of the particles, ca. 90% of the supernatant was decanted and the remaining material was centrifuged for 5 min whereupon the acetone layer was removed, allowing the colloidal precipitate to be re-suspended as necessary.
000; PVP/Pd ratio ca. 10:1) was dissolved in 120 ml of anhydrous ethylene glycol and the resulting solution was heated. After adjusting the pH to 9–11 (ca. 5 ml of 1 M NaOH), the palladium acetate solution was added and the reaction was stirred to yield a black colloidal suspension (batch 1, 100 °C for 1 h; batch 2, 70 °C for 2 h). Aliquots of the product were purified by extracting 50 ml suspensions from ethylene glycol and surplus polymer using excess acetone. After sedimentation of the particles, ca. 90% of the supernatant was decanted and the remaining material was centrifuged for 5 min whereupon the acetone layer was removed, allowing the colloidal precipitate to be re-suspended as necessary.
2.2 Fabrication of ZnO nanowires
 | ||
| Fig. 2 Schematic diagram of a CVD apparatus used to grow ZnO nanowires onto Si substrates bearing a ZnO seed layer. | ||
2.3 Pd nanoparticles coated ZnO nanowires
The ZnO nanowires were subsequently spin-coated at 1500 rpm with two different ethanol solutions containing Pd nanoparticles20 with an average size of 2.7 (Pd batch 2) and 4.5 nm (Pd batch 1). Quantities were calculated to give 1wt% Pd/ZnO assuming complete uptake of the noble metal. The substrates were dried and calcined at 773 K under air for 2 h. The resulting samples are referred to as Pd(4.5)/ZnO and Pd(2.7)/ZnO, where (4.5) and (2.7) indicate the size of the Pd nanoparticles.2.4 Characterization
The crystal structure and texture of the ZnO thin films (i.e. seed layers) and the crystalline structure of the NWs were characterized by X-ray diffraction (Bruker, Cu-Kα radiation with wavelength of 1.5406 Å). The thickness of the ZnO films was determined from the cross-sectional SEM images. The microstructures of the ZnO seed layers and NWs as well as the supported nanoparticle size distributions were examined using a combination of scanning electron microscopy (SEM, Philips XL30 ESEM-FEG) and transmission electron microscopy (TEM). TEM micrographs were obtained using a JEM-2010 microscope at 200 kV with a resolution of 0.14 nm. Samples for TEM were prepared by placing a slice of the film removed from the substrate on a copper grid coated with a carbon film and dried under vacuum. The surface morphology of the films was examined using SEM. The XPS measurements of Pd(4.5)/ZnO NWs were carried out with a Kratos AXIS Ultra spectrometer, equipped with a monochromatic Al Kα X-ray source and a delay-line detector (DLD). Spectra were obtained using an aluminium anode (Al Kα = 1486.6 eV) operating at 150 W. The background pressure was 2 × 10−9 mbar.2.5 Hydrogenation of 2-methyl-3-butyn-2-ol
The ZnO NWs or Pd/ZnO NWs films were placed in an autoclave reactor with a total volume of 270 ml and were reduced in situ at 523 K at a hydrogen pressure of 5 bar for 6 h. Then, the reactor was cooled down to room temperature. A 6 mM solution of C5H7OH (MBY) in methanol, water or methanol-water (75–25 vol.%) mixture (130 ml) was deoxygenated and transferred into the reactor. Analysis of the reaction mixture was performed on-line by introducing aliquots at established time intervals into a Varian CP-3800 GC equipped with a CP-Sil 5 CB capillary column. The carbon balance was closed to 99%. No C10-dimer formation was observed. The turnover frequency (TOF) was calculated as follows:where X is the MBY conversion, NMBY is the amount of MBY (mol), mcat is the catalyst amount (g), α is Pdloading, MPd is atomic weight of Pd (g mol−1), t the reaction time (s), and D the Pd dispersion. For the average Pd particle size as determined using TEM, the Pd dispersion was estimated using the following equation: D ≈ 0.9/dTEM (nm).21

3. Results and discussion
3.1 Characterization of ZnO and Pd/ZnO NWs catalysts
XRD results revealed that ZnO thin films (i.e. seed layers) were polycrystalline with a hexagonal wurtzite structure (Fig. 3c). These ALD deposited ZnO thin films grown at 553 K were highly textured (00.2) with the c-axis perpendicular to the substrate surface. This is evident from Fig. 3a where no diffraction is observed in the (10.0) pole figure of the film deposited at 553 K, whereas very strong diffraction intensity is seen in the middle of the (00.2) pole figure (see Fig. 3b). | ||
| Fig. 3 Pole figures of XRD texture analysis on ZnO seed layer deposited by ALD on Si (100) at 553 K (a) (10.0) plane; (b) (00.2) plane; (c) XRD trace of ZnO thin films (i.e. seed layers) grown by ALD at 553 K. The nanoparticle size is shown in nanometers. | ||
The XRD pattern for the undecorated ZnO NWs is shown in Fig. 4. The structure corresponds to a zincite hexagonal P63mc structure (cell parameters of a = 3.2 Å, c = 5.2 Å), with a small shift to higher 2θ values.22
 | ||
| Fig. 4 XRD pattern of ZnO NWs. | ||
SEM and TEM images of the as-synthesized ZnO NWs are shown in Fig. 5a and b. The average length ranges from 1.5 to 10 μm and diameter in the range of 144 ± 8 nm. The overall average areal density of the ZnO NWs is 18.2 ± 0.5 NWs/μm2. The ZnO NWs grown on highly textured seed layers are vertically aligned (Fig. 5a). TEM images of the Pd(2.7)/ZnO and Pd(4.5)/ZnO samples are shown in Fig. 5c and d, respectively. Both catalysts contain monodispered Pd nanoparticles with an average diameter of 4.5 ± 0.1 and 2.7 ± 0.1 nm which are homogeneously distributed over the ZnO NWs surface. The particle size distribution plots are shown in Fig. 6.
 | ||
| Fig. 5 (a) SEM image of ZnO NWs; (b) TEM images of ZnO NWs, (c) Pd(2.7)/ZnO NWs and (d) Pd(4.5)/ZnO NWs. | ||
 | ||
| Fig. 6 Pd particles size distribution for (a) Pd(4.5)/ZnO NWs and (b) Pd(2.7)/ZnO NWs. 100 particles were counted in each case. The nanoparticle size is shown in nanometers. | ||
Pd 3d XPS spectra of fresh and spent (after 2 catalytic cycles in methanol, vide infra) Pd(4.5)/ZnO NWs sample are shown in Fig. 7 (a and b, respectively). In both cases XPS peaks appeared at 335.2, 336.7 and 338.0 eV. These values are characteristic of Pd-species with mixed valences: Pd(0), Pd(II) and Pd(IV).23
 | ||
| Fig. 7 XPS data for Pd(4.5)/ZnO NWs: (a) before catalyst testing, (b) after 2 runs of the reaction with methanol as a solvent. | ||
3.2 MBY hydrogenation
:1 vol.) mixture. The lowest activity and a selectivity of 76% towards MBE were observed when water was used (Table 1). The Pd particle size increased from 2.7 ± 0.1 to 3.3 ± 0.3 nm after two experimental runs (Fig. 8b and c). Replacement of water with methanol increased the selectivity towards the semihydrogenated product. Higher MBY selectivities, of 82 and 88%, were observed in methanol-water mixture and pure methanol, respectively. The activity of the Pd(2.7)/ZnO catalyst remained constant over two hydrogenation runs. TEM analysis (Fig. 8a) showed that the diameter of the ZnO nanowires remained the same, but the Pd nanoparticle diameter increased from 2.7 ± 0.1 to 3.5 ± 0.2 nm in the methanol-water mixture. In pure methanol, the Pd particle diameter of 2.7 ± 0.1 nm remained unchanged after three hydrogenation runs.
| Pressure, bara | Solvent | Initial TOF, s−1 | Selectivity to MBEa (%) | Average diameter of Pd particles, nm |
|---|---|---|---|---|
| Reaction temperature during all runs was kept at 303 K.a Values are given at 99.9% MBY conversion.b Measured before 1st run in water.c Measured after 2nd run in water.d Measured after 2nd run in water–methanol.e Measured after 3rd run in methanol. n.d. = not determined. | ||||
| 5 | H2O | 1.5 | 77 ± 2 | 2.7 ± 0.1b |
| 5 | H2O | 1.3 | 75 ± 2 | 3.3 ± 0.3c |
| 5 | 25%vol. H2O in MeOH | 3.1 | 82 ± 2 | n.d. |
| 5 | 25%vol. H2O in MeOH | 2.9 | 82 ± 2 | 3.5 ± 0.2d |
| 5 | MeOH | 1.5 | 88 ± 2 | n.d. |
| 5 | MeOH | 3.2 | 86 ± 2 | n.d. |
| 2 | MeOH | 3.2 | 88 ± 2 | 2.7 ± 0.1e |
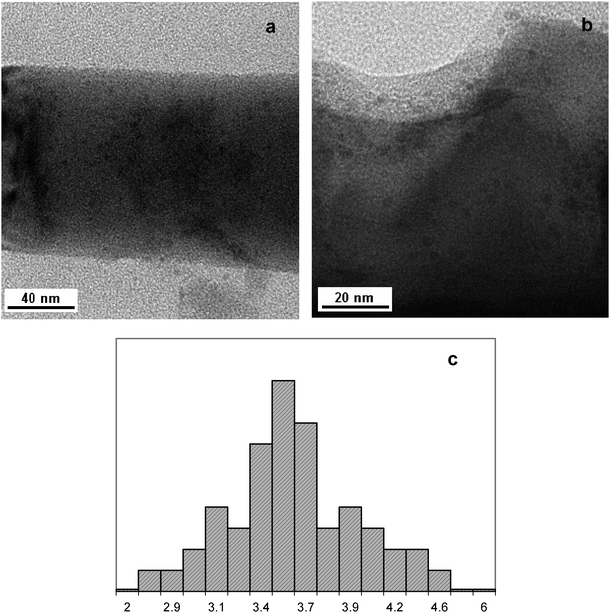 | ||
| Fig. 8 TEM image of Pd(2.7)/ZnO NWs after testing in (a) methanol-water as a solvent (2 runs); (b) water as a solvent (1 run); (c) Pd particle size distribution for the Pd(2.7)/ZnO NWs after 2 runs of the reaction with methanol-water as a solvent (50 particles were counted). The nanoparticle size is shown in nanometers. | ||
The reaction scheme of the MBY hydrogenation is shown in Fig. 1. There exist three pathways in the MBY hydrogenation reaction. The first one is a conversion of MBY to the semihydrogenated product (MBE), the second one takes place through the conversion of MBE to the fully hydrogenated product (MBA), and the third one is the direct alkyne-to-alkane hydrogenation reaction. Fitting functions (eqn (1)–(3)) can be used to describe the experimental results:
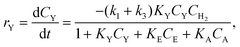 | (1) |
 | (2) |
 | (3) |
The concentrations of MBY, MBE, and MBA as a function of time are shown in Fig. 9. The kinetic parameters were obtained by using regression analysis to minimize the residual sum of squares between the experimental and calculated concentrations at the reactor outlet. All experimental data were treated with equal statistical weights. The model data (solid lines) reveal an excellent match with the experimental data (circular symbols). The kinetic parameters are presented in Table 2.
 | ||
| Fig. 9 Product distribution during the MBY hydrogenation reaction on Pd(2.7)/ZnO NWs using different solvents: (a) water (1st run); (b) 25%vol. water in methanol (1st run); (c) methanol (2nd run). Solid lines represent the concentration profiles obtained by fitting the kinetic model, symbols represent experimental data. Reaction conditions: 6 mM solution of MBY, hydrogen pressure 5 bar, temperature 303 K. | ||
| Kinetic parameters | Water 1st run | 25%vol. water in methanol 1st run | Methanol 2nd run |
|---|---|---|---|
| k 1, m3 mol−1 min−1 | 2.3 × 10−3 ± 3 × 10−4 | 2.6 × 10−3 ± 2 × 10−4 | 2.8 × 10−3 ± 2 × 10−4 |
| k 2, m3 mol−1 min−1 | 3.2 × 10−4 ± 5 × 10−5 | 3.3 × 10−4 ± 5 × 10−5 | 8.7 × 10−5 ± 5 × 10−6 |
| k 3, m3 mol−1 min−1 | 6.5 × 10−4 ± 3 × 10−5 | 7.1 × 10−4 ± 2 × 10−5 | 3.4 × 10−4 ± 2 × 10−5 |
| K Y, m3 mol−1 | 2.1 × 103 | 2.5 × 103 | 5.1 × 103 |
| K E, m3 mol−1 | 0.8 | 0.45 | 2.0 × 10−2 |
| K A, m3 mol−1 | 1.3 × 10−2 (fixed) | 1.3 × 10−2 (fixed) | 1.3 × 10−2 (fixed) |
| K Y/KE, – | 2.6 × 103 | 5.6 × 103 | 2.6 × 105 |
| k 1/k3, – | 3.5 | 3.7 | 8.2 |
In the case of using methanol and the methanol-water mixture, the TOF was found to be of the same value (ca. 3.2 and 3.0, respectively). As compared with the methanol-water mixture, the rate constant k1 was 10% higher in methanol and it was 10% lower in water. This can be explained by the different polarities of the solvents. The coordination of the adsorbed species is influenced by their environment which is different in methanol and water and it was reported in25 that the hydrogenation activity decreases with increasing solvent polarity due to competitive adsorption of the polar solvent and the reagent on the catalyst surface.
The fully hydrogenated product was produced from the beginning of the reaction in all three solvents. The increase of the selectivity towards MBE in the following sequence—water < methanol–water < methanol—is in agreement with an increase in the k1/k3 ratio (Table 2). The KY/KE ratio steadily increases from 2.6 × 103 to 2.6 × 105 for water and methanol, respectively. In the case of methanolKY is twice higher than in water, however KE is 40 times lower. In the case of methanol-water mixture both KY and KE possess intermediate values. The selectivity to MBE increases after substitution of water with methanol also because the direct MBY–to–MBA hydrogenation is suppressed. This fact is confirmed by the increase of the k1/k3 ratio: 3.5, 3.7 and 8.2 for water, water–methanol and methanol, respectively. The increase of the rate of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond hydrogenation using the polar solvents was observed therein.25–27 One of the possible explanations is that the polar solvents favour adsorption of double bond containing compounds on the catalyst surface, because of their lower affinity for this molecule.25,26 However, it has been mentioned that several factors can be responsible for the effect of solvent on the activity and selectivity, e.g. solubility of the liquid and gaseous reactants and their adsorption on the catalyst surface, competitive adsorption of solvent molecules, interaction of the solvent with the reactant(s) either in the liquid-phase or on the catalyst surface as well as catalyst deactivation caused by the solvent. Therefore, the possible solvent effects manifest here combine very different physical and chemical phenomena and thus it is not straightforward to present a complete picture.28
C bond hydrogenation using the polar solvents was observed therein.25–27 One of the possible explanations is that the polar solvents favour adsorption of double bond containing compounds on the catalyst surface, because of their lower affinity for this molecule.25,26 However, it has been mentioned that several factors can be responsible for the effect of solvent on the activity and selectivity, e.g. solubility of the liquid and gaseous reactants and their adsorption on the catalyst surface, competitive adsorption of solvent molecules, interaction of the solvent with the reactant(s) either in the liquid-phase or on the catalyst surface as well as catalyst deactivation caused by the solvent. Therefore, the possible solvent effects manifest here combine very different physical and chemical phenomena and thus it is not straightforward to present a complete picture.28
The selectivity and activity of the Pd(4.5)/ZnO NWs catalyst decrease after the second hydrogenation run (Table 3) because of a deactivation process that can be related to the deposition of organic species on the noble metal surface and sintering of Pd nanoparticles. These two phenomena can reasonably be viewed as causing the partially reversible deactivation of the catalyst. After regeneration at 573 K for 2 h, the catalyst showed activity and MBE selectivity that was higher than that seen in the second hydrogenation run, but still lower than that in the first run. Both activity and selectivity dropped because of the aggregation of Pd nanoparticles. The regeneration at higher temperatures led to further catalyst deactivation. The Pd nanoparticle size increased to 7.5 and 9 nm after calcination at 773 and 823 K, respectively. Such significant sintering can be explained by complete PVP removal above 773 K.
| Before reaction | 1st run | 2nd run | After regeneration | |||
|---|---|---|---|---|---|---|
| 573 K/2 h/air | 773 K/2 h/air | 823 K/2 h/air | ||||
| a Values are given at 99.9% MBY conversion. | ||||||
| Pd particles size, nm | 4.5 ± 0.1 | n.d. | n.d. | n.d. | 7.5 ± 0.2 | 9.0 ± 0.3 |
| Initial TOF, s−1 | — | 0.59 | 0.48 | 0.53 | 0.27 | 0.04 |
| MBE selectivitya (%) | — | 95 ± 2 | 91 ± 2 | 91 ± 2 | 87 ± 2 | 86 ± 2 |
Blank experiments were performed with ZnO NWs. The MBE selectivity over these ZnO NWs was rather low at 313 K (Fig. 10). As the temperature was increased, both the reaction rate and the MBE selectivity increased. The highest selectivity of 89% was observed at 333 K and 5 bar H2 pressure. Further increases in temperature resulted in a decrease in the selectivity to 83% at 343 K. Nevertheless, in our view the selectivity of 89% towards MBE can be considered as rather high in the absence of noble metal. Indeed, there is still a chance to increase the selectivity by optimizing the solvent and the temperature. Furthermore, the ZnO NWs catalyst was observed to be stable, with no deactivation observed after 20 hydrogenation cycles.
 | ||
| Fig. 10 Selectivity towards MBE using ZnO NWs catalyst under different reaction conditions. Reaction conditions: 6 mM solution of MBY in methanol, hydrogen pressure 3–10 bar, temperature 313–343 K. | ||
The concentrations of MBY, MBE, and MBA as a function of time are shown in Fig. 11a, b and c for the first experimental run using a ZnO NWs sample, and the first and second runs using the Pd(4.5)/ZnO NWs catalyst, respectively. The model data (solid lines) is in good agreement with the experimental data (circular symbols). The kinetic parameters are listed in Table 4.
 | ||
| Fig. 11 Product distribution during the MBY hydrogenation reaction on: (a) ZnO NWs; (b) Pd(4.5)/ZnO NWs (1st run); (c) Pd(4.5)/ZnO NWs (2nd run). Solid lines represent the concentration profiles obtained by fitting the kinetic model, symbols represent experimental data. Reaction conditions: 6 mM solution of MBY in methanol, hydrogen pressure 5 bar, temperature 323 K. | ||
| Kinetic parameters | ZnO NWs | Pd(4.5)/ZnO NWs 1st run | Pd(4.5)/ZnO NWs 2nd run |
|---|---|---|---|
| k 1, m3 mol−1 min−1 | 2.53 × 10−4 ± 5 × 10−6 | 2.61 × 10−4 ± 2 × 10−6 | 2.38 × 10−4 ± 2 × 10−6 |
| k 2, m3 mol−1 min−1 | 1.0 × 10−4 ± 8 × 10−5 | 7 × 10−4 ± 1 × 10−4 | 3.4 × 10−4 ± 1 × 10−5 |
| k 3, m3 mol−1 min−1 | 4.3 × 10−5 ± 4 × 10−6 | 1.6 × 10−5 ± 2 × 10−6 | 3.8 × 10−5 ± 1 × 10−6 |
| K Y, m3 mol−1 | 20 | 7.7 × 103 | 2.4 × 104 |
| K E, m3 mol−1 | 2.5 × 10−2 | 2.3 × 10−2 | 2.3 × 10−2 |
| K A, m3 mol−1 | 1.3 × 10−2 (fixed) | 1.3 × 10−2 (fixed) | 1.3 × 10−2 (fixed) |
| K Y/KE, – | 8.0 × 102 | 3.4 × 105 | 1.1 × 106 |
| k 1/k3, – | 5.9 | 16.2 | 6.2 |
| k 2/k3, – | 2.3 | 46.3 | 8.8 |
In the case of Pd(4.5)/ZnO NWs, 99.5% conversion of MBY was achieved after 22 and 24 h during the first and second runs, respectively. The rate constant k1 decreased by 9% during the second run, and this likely indicates the onset of catalyst deactivation. Alkane was produced from the beginning of the reaction. The selectivity towards MBE at 99.9% MBY conversion decreased from 95 to 91%. This observation is in agreement with a 2.6-fold decrease in the k1/k3 ratio in the second run as compared with the first run. In the case of the ZnO NWs the selectivity towards MBE was found to be 89% and the k1/k3 ratio was calculated to be 5.9.
The KY/KE ratio of 8.0 × 102 for the ZnO NWs was of the same order of magnitude as has been reported previously.6,30 In contrast, the KY/KE ratios of 3.4 × 105 and 1.1 × 106 observed presently for cycles 1 and 2 using Pd(4.5)/ZnO are 2–4 orders of magnitude higher than those reported elsewhere6,30 for the hydrogenation of MBY and phenylacetylene on Pd25Zn75/TiO2 and CNF/Ti/TiO2 catalysts. Since KE was of the same magnitude as reported previously,6 this can be taken as being indicative of the stronger adsorption of MBY on the Pd(4.5)/ZnO NW surface.
During the first experimental cycle using Pd(4.5)/ZnO, MBE formation only became significant after MBY was consumed, whereas during the second cycle the parallel mechanism makes a greater contribution to the formation of MBA (k2/k3 ratios are 46.3 and 8.8 for the first and second runs, respectively). This observation was in agreement with data obtained previously during the hydrogenation of phenylacetylene.30 In the case of ZnO NWs, the influence of the direct alkyne–to–alkane hydrogenation was even higher (k2/k3 ratio is 2.3). During the first experimental run deploying Pd/ZnO the selectivity towards MBE dropped from 97 to 95% during the first hour before remaining constant until MBY had undergone complete conversion. In the second run, the selectivity towards MBE decreased from 94 to 91% over the first 3 h, remaining constant thereafter. In the case of ZnO NWs the selectivity towards MBE (89%) did not decrease until the MBY substrate was completely consumed.
Pd(2.7)/ZnO NWs catalysts were tested in MBY hydrogenation at a temperature of 303 K and at different hydrogen pressures (2 and 5 bar). The selectivity towards MBE was found to be 86–88% each of the three times that the reaction was run. The activity of the catalyst (TOF) exhibited a 2-fold increase during the second run of the reaction (Table 1) and remained the same during the third run. The Pd(2.7)/ZnO NWs catalysts exhibited better activity but lower selectivity in comparison with the Pd(4.5)/ZnO NWs catalysts. However, as can be seen from Table 3, for the Pd(4.5)/ZnO NWs catalyst the selectivity dropped from 95 to 91% during the second run of the reaction because of catalyst deactivation. The lower selectivity and absence of deactivation in the case of Pd(2.7)/ZnO NWs can be explained by the lower temperature at which the reaction was conducted (303 K). Based on our observations, the higher activity in MBY hydrogenation can clearly be related to the use of smaller Pd nanoparticles.
In comparing the present results with previously reported literature data (Table 5) the following advantages of using Pd/ZnO NWs catalysts can be mentioned: no separation of the catalyst from reaction mixture is needed; there is a possibility of using less toxic solvents (water instead of methanol) and this has no significant influence on the ZnO NWs structure; these NWs exhibit a highly porous and easily accessible structure, which increases the effective diffusivity of large organic molecules into the porous domain.
| Catalyst | Selectivity towards MBEa (%) | Remarks | Ref. |
|---|---|---|---|
| a At 99% MBA conversion. | |||
| CNF/Ti/TiO2 | 73–86 | Relatively low selectivity. | 30 |
| Pd25Zn75/TiO2 wall-coated capillary | 97 | Pyridine was added to the system → additional separation step from the reaction mixture; very low amount of catalyst → not effective use of the reactor volume. | 6 |
| Pd/ZnO on sintered metal fibers | 95.5 | Quinoline was added to the system → additional separation step from the reaction mixture needed. | 31 |
| Monodispersed Pd nanoparticles | 96 | Batch process → separation of the catalyst from the reaction mixture needed. | 23 |
| ZnO NWs and Pd/ZnO NWs | 77–95 | No separation of the catalyst from the reaction mixture needed; possibility of using less toxic solvents. | Present work |
Conclusions
Vertically aligned ZnO NWs have been successfully grown by CVD onto highly (00.2) textured ZnO seed layers deposited by ALD at 553 K. The ZnO nanowires were subsequently spin-coated with Pd nanoparticles, and the resulting catalysts were tested in the hydrogenation of 2-methyl-3-butyn-2-ol in a batch reactor. The ZnO NWs showed no deactivation after 20 hydrogenation cycles at 323 K. The MBE selectivity of 95% at an alkyne conversion of 99% was observed over the Pd(4.5)/ZnO catalyst at 323 K. Catalytic tests performed in water and in a methanol–water (3:1 vol.) mixture led to sintering of the Pd particles, decreasing both the activity and selectivity. The highest TOF of 3.2 s−1 and selectivity of 88% were observed over the Pd(2.7)/ZnO NWs catalyst in methanol. This catalyst remains stable during three subsequent hydrogenation runs. In all cases, the ZnO NWs proved to be rather stable in water, which is promising for further optimization of their activity and stability using highly polar solvents.
Acknowledgements
The authors thank Dr M. W. G. M. Verhoeven and Mr S. Çelebi from Eindhoven University of Technology for XPS and TEM measurements. The financial support by the Netherlands Organisation for Scientific Research (NWO), the Russian Foundation for Basic Research (RFBR) in the frame of NWO-RFBR project 047.017.028, and Royal Society International joint project 2008/R4 “Smart Structured Multiphase Reactors for Process Intensification” is gratefully acknowledged.References
- S. J. Lim, S. Kwon and H. Kim, Thin Solid Films, 2008, 516, 1523 CrossRef CAS.
- S. Y. Pung, K. L. Choy, X. Hou and C. Shan, Nanotechnology, 2008, 19, 435609 CrossRef.
- S. C. Su, Y. M. Lu, Z. Z. Zhang, B. H. Li, D. Z. Shen, B. Yao, J. Y. Zhang, D. X. Zhao and X. W. Fan, Phys. B, 2008, 403, 2590 Search PubMed.
- Z. L. Wang, Mater. Sci. Eng., R, 2009, 64, 33 CrossRef.
- N. Semagina, M. Grasemann, N. Xanthopoulos, A. Renken and L. Kiwi-Minsker, J. Catal., 2007, 251, 213 Search PubMed.
- E. V. Rebrov, E. A. Klinger, A. Berenguer-Murcia, E. M. Sulman and J. C. Schouten, Org. Process Res. Dev., 2009, 13, 991 Search PubMed.
- N. Semagina, A. Renken, D. Laub and L. Kiwi-Minsker, J. Catal., 2007, 246, 308 CrossRef CAS.
- E. Sulman, V. Matveeva, N. Semagina, I. Yanov, V. Bashilov and V. Sokolov, J. Mol. Catal. A: Chem., 1999, 146, 257 CrossRef CAS.
- US Patent, 3 696 155, 1972 Search PubMed.
- J. M. Planeix, N. Coustel, B. Coq, V. Brotons, P. S. Kumbhar, R. Dutartre, P. Geneste, P. Bernier and P. M. Ajayan, J. Am. Chem. Soc., 1994, 116, 7935 CrossRef CAS.
- M. J. F. Warnier, PhD thesis, Eindhoven University of Technology, 2009 Search PubMed.
- E. V. Ramos-Fernández, A. F. P. Ferreira, A. Sepúlveda-Escribano, F. Kapteijn and F. Rodríguez-Reinoso, J. Catal., 2008, 258, 52 Search PubMed.
- P. S. Wehner and B. L. Gustafson, J. Catal., 1992, 135, 420 Search PubMed.
- X.-L. Liang, X. Dong, G.-D. Lin and H.-Bi. Zhang, Appl. Catal., B, 2009, 88, 315 CrossRef CAS.
- M. Consonni, D. Jokic, D. Yu. Murzin and R. Touroude, J. Catal., 1999, 188, 165 CrossRef CAS.
- S. K. Spear, S. T. Griffin, K. S. Granger, J. G. Huddleston and R. D. Rogers, Green Chem., 2007, 9, 1008 RSC.
- C. Badre, T. Pauportel, M. Turmine and D. Lincot, Nanotechnology, 2007, 18, 365705 CrossRef.
- X. Tang, E. S. G. Choo, L. Li, J. Ding and J. Xu, Langmuir, 2009, 25(9), 5271 CrossRef CAS.
- K. L. Choy, Prog. Mater. Sci., 2003, 48(2), 57 CrossRef CAS.
- S. Domínguez-Domínguez, A. Berenguer-Murcia, D. Cazorla-Amorós and A. Linares-Solano, J. Catal., 2006, 243, 74 CrossRef CAS.
- M. Boudart, in Kinetics of Heterogeneous Catalytic Reactions, Princeton Univ. Press, Princeton, NJ, 1984, p. 26 Search PubMed.
- H. F. McMurdie, F. Howard, M. C. Morris, E. H. Evans, B. Paretzkin, W. Wong-Ng, L. Ettinger and C. R. Hubbard, Powder Diffr., 1986, 1, 76 Search PubMed.
- N. Semagina, A. Renken, D. Laub and L. Kiwi-Minsker, J. Catal., 2007, 246, 308 CrossRef CAS.
- C. Descamps, C. Coquelet, C. Bouallou and D. Richon, Thermochim. Acta, 2005, 430, 1 CrossRef CAS.
- M. Khodadadi-Moghaddama, A. Habibi-Yangjehb and M. Reza Gholami, J. Mol. Catal. A: Chem., 2009, 306, 11 CrossRef CAS.
- J. L. Dallons, I. G. Jannes and B. Delmon, Catal. Today, 1989, 5, 257 Search PubMed.
- L. Cerneny, J. Vopatova and V. Ruzicka, React. Kinet. Catal. Lett., 1982, 19, 223 Search PubMed.
- P. G. J. Koopman, H. M. A. Buurmans, A. P. G. Kieboom, H. van Bekkum and J. R. Recueil, Neth. Chem. Soc., 1981, 100, 156 Search PubMed.
- D. V. Sokolskii, N. V. Anisimova, A. K. Zharmagarnbetova and A. Ualikhanova, React. Kinet. Catal. Lett., 1981, 17, 419 Search PubMed.
- A. Berenguer-Murcia, E. V. Rebrov, M. Cabaj, A. E. H. Wheatley, B. F. G. Johnson, J. Robertson and J. C. Schouten, J. Mater. Sci., 2009, 44, 6563 Search PubMed.
- N. Semagina, M. Grasemann, N. Xanthopoulos, A. Renken and L. Kiwi-Minsker, J. Catal., 2007, 251, 213 Search PubMed.
Footnote |
| † Present address: School of Chemistry and Chemical Engineering, Queen's University Belfast, Stranmillis Road, Belfast, BT9 5AG, UK |
| This journal is © The Royal Society of Chemistry 2011 |