Metal–organic frameworks as heterogeneous catalysts for oxidation reactions
Amarajothi
Dhakshinamoorthy
,
Mercedes
Alvaro
and
Hermenegildo
Garcia
*
Instituto Universitario de Tecnología Química CSIC-UPV and Departamento de Química, Universidad Politécnica de Valencia, Av. De los Naranjos s/n, 46022 Valencia, Spain. E-mail: hgarcia@qim.upv.es; Fax: +34 9638 7807
First published on 28th April 2011
Abstract
In this Perspective, we describe the use of metal–organic frameworks (MOFs) as heterogeneous catalysts for oxidations using hydroperoxides or molecular oxygen. These two types of oxidants fulfill the requirements of green chemistry in terms of environmental benignity and sustainability. For the sake of clarity and to illustrate the possibilities of using MOFs as oxidation catalysts, we have constrained ourselves to present the results obtained for the oxidation of cycloalkanes, oxidation of benzylic positions, oxidation of cycloalkenes and aerobic oxidation of alcohols. The use of MOFs as catalysts for enantioselective oxidations has been dealt with in a separate section in which the peculiarity of the required oxidants and the advantages of having homo chiral MOFs have been discussed. The key features of MOFs as catalysts, the similarities with inorganic porous solids and future developments in this field have been discussed in separate sections.
1. Introduction
Metal–organic frameworks (MOFs) are crystalline porous materials whose structure is defined by metal ions or clusters of metals that are connected to bi or tripodal rigid organic linkers.1–4 The directionality of metal–ligand interaction and the angles defined by the multipodal linker determine the formation of cavities and channels of nanometric dimensions that can be accessed from the exterior allowing mass transfer and the incorporation of substrates inside the crystal.3 MOFs are characterized by being at the top of the list of the porous materials with the largest surface area, lowest framework density and higher pore volume.5 As an example, Fig. 1 shows a general MOF structure.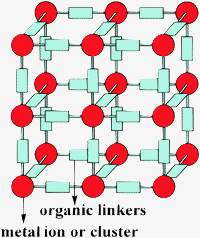 | ||
| Fig. 1 General structure of MOFs. | ||
One additional characteristic of MOFs is that sometimes it is possible to predict the crystal structure of new materials based on the knowledge of metal clusters, their preferred directionalization and geometry and dimensions of the organic linkers.6,7 To illustrate this point Fig. 2 shows how a series of aromatic dicarboxylates can be used as linkers for the synthesis of isomorphic MOFs with identical crystal structure but with increasing dimensions of unit cell.
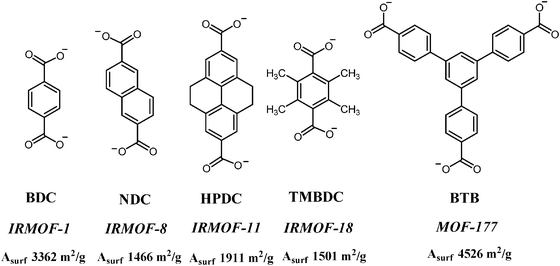 | ||
| Fig. 2 Aromatic dicarboxylates varying in length used as linkers for a series of isoreticular MOFs. The common acronym of the linker and MOF as well as the resulting surface area have also been indicated. | ||
Beyond the synthesis of new materials, the interest in MOFs derives from the large porosity and their potential use as adsorbents for hydrogen, carbon dioxide and other gases.8–14 It has also been reported that MOFs can be used for the separation and sequestration of some molecules present in the gas phase.15Scheme 1 shows the expected possible applications of MOFs in different sectors.
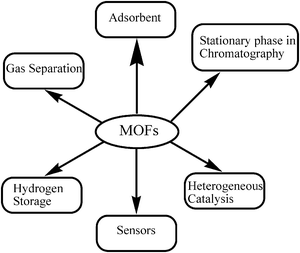 | ||
| Scheme 1 Possible applications of MOFs in various areas. | ||
MOFs versuszeolites
MOFs have crystal structures that are related to those of zeolites and other inorganic porous solids. Considering the high metal content in MOFs and their similarity with zeolites, these materials are attracting an increasing interest as heterogeneous catalysts.16–28Table 1 provides a comparison of some features that are relevant in catalysis for zeolites and MOFs.| Properties | Zeolites | MOFs |
|---|---|---|
| Lewis acidity | High | Low to medium |
| Brønsted acidity | Bridging Si(OH)/Al hydroxyl groups | Introduced by post-synthetic modifications or functional groups present in the ligands not compromised in the structure |
| Surface area | Around 200–500 m2 g−1 | Up to 5000 m2 g−1 |
| Pore volume | Around 0.1–0.5 cm3 g−1 | Over 1 cm3 g−1 |
| Thermal stability | >450 °C | 300 °C |
| Chemical stability | High | Limited and incompatible with certain solvents |
| Diffusion | High for small molecules in the gas phase and slow in the liquid phase | Strongly influenced by the polarity of linkers |
| Basicity | Arises from the framework oxygens | Introduced by post-synthetic modifications or directly through linkers |
| Metal site density | Low | High |
| Framework defects | Plays an important role in many reactions | Expected to play a minor role |
| Active site environment | Mostly hydrophilic but can also be made hydrophobic | Considerably hydrophobic |
| Poisoning of active sites | Reactivation by thermal treatment | Thermal treatment not valid for catalyst regeneration |
| Chirality | Not possible or difficult to achieve | Homochiral solids can be easily obtained from chiral linkers or postsynthetic modifications |
As it can be seen there, according to our view, zeolites are much more suited as solid catalysts for gas phase reactions but due to their limited available pore size they generally undergo fast deactivation in liquid phase reactions. In addition most of the efforts to develop zeolites with large pore size in the nanometre scale have met with failure.29,30 In liquid phase reactions, diffusion can be a controlling process and also considering the larger molecular dimensions of substrates that can be employed in a liquid phase process, it would be highly important to have porous catalysts with pore dimensions larger than those of classical zeolites.
In this regard, MOFs can be considered as complementing in expanding the work of zeolites in heterogeneous catalysis. Certainly, due to their low thermal stability, MOFs are not likely to be of potential interest for gas phase reactions. On the contrary the large versatility in the design and engineering of MOFs with adequate geometry and pore size makes these materials more promising for liquid phase reactions, just the type of transformations for which the performance of zeolites is generally not adequate. Although there are some scattered examples for which zeolites are excellent solid catalysts in the liquid phase such as selective hydrogenation of trans-fatty acids on the Pt-ZSM-5 zeolite31 or selective macrolactonation of hydroxycarboxylic acids on HY zeolites,32 there are many other cases in which zeolites become immediately deactivated due to poisoning when the reaction is carried out in the liquid phase.
Finally, one field in which MOFs are certainly much better materials than zeolites is in promoting enantioselective reactions. Using chiral linkers, preparation of homochiral MOFs can be easily obtained and these solids could be suitable as catalysts for asymmetric reactions.33 Besides hydrogenation, asymmetric oxidation is the field that has attracted considerable attention since the resulting products and, particularly, chiral epoxides are very important synthetic intermediates in asymmetric reactions.34
Stability of MOFs
In the present perspective, we comment on the use of MOFs as catalysts for liquid-phase oxidations. As in any use of MOFs in catalysis, the key points to be addressed are their activity, selectivity, stability under the reaction conditions and deactivation pathways. Thus, in addition to providing conversion and selectivity data, when using MOFs it is also necessary to determine the stability of the material and the lack of leaching of active metal species to the solution. These points are typically addressed by checking the crystal structure and determining the surface area after the catalytic reactions as well as performing analysis of the solutions at final reaction time. Productivity, defined as the amount of product that can be obtained with a given amount of catalyst before deactivation, is also one of the important data that must be considered in order to assess the feasibility of using MOFs as catalysts.Concerning the use of MOFs as oxidation catalysts, there is a considerable interest in green chemistry to replace stoichiometric oxidations using transition metals or other environmentally unfriendly oxidants by environmentally benign ones.20 The ultimate green oxidants in terms of the percentage of active oxygen and toxicity of the byproducts are hydrogen peroxide, organic hydroperoxides and oxygen.35Oxidations using peroxides and oxygen require the use of a suitable catalyst and those based on transition metals are of general use. While metal oxides and other inorganic compounds are non-porous, the catalytic activity per metal atom (turnover number, TON) tends to be low and in order to increase these TON values a reduction of particle size down to the nanometre scale is necessary. Even for nanoparticulate materials, it would be convenient to have all the metal sites exposed on the surface of the solid. This strategy to isolate transition metal atoms on a large surface area solid has been developed by having transition metal containing zeolites and using them as oxidation catalysts.36 The activity of these metal containing zeolites for many oxidations particularly in the gas phase has been found to be very high and they have been used in several industrial processes.37,38 However, one disadvantage of transition metal containing zeolites is the low metal content that can be achieved. This limitation can be overcome using MOFs as catalysts. Since these solids can have a weight percentage of transition metals of over 20% (typically zeolites contain only a few percentage of transition metals), these metals are dispersed in a very large surface area.
One point to be addressed in any heterogeneous catalyst, but also of particular relevance in MOFs, is the stability of the catalyst and in particular the absence of metal leaching. For instance in the case of transition metal containing zeolites metal leaching under optimum conditions in the presence of oxidizing agents is a general problem that has to be carefully surveyed and in the case that the total absence of leaching is proposed it has to be convincingly proved. A strong oxidant generates stress in the catalytic sites since the mechanism can require either reversible changes in the oxidation state or a cycle of ligand exchange. Therefore going from one step to the other in the mechanism typically requires changes in the coordination sphere in the metal ion and these changes can be large enough to effect the migration of the metal out of the solid framework. In the case of MOFs, one additional problem is the stability of the organic linker to the oxidative stress and the strength of the metal-cluster linker interaction that could be not strong enough to completely avoid metal leaching under the typical conditions and solvents used in oxidation reactions.
Suitable oxidizing reagents
One of the principles of Green Chemistry is the replacement of stoichiometric by catalytic reactions.39Oxidation reaction is a typical case in which for environmental reasons the change of stoichiometric oxidants for a catalytic process is particularly important due to the toxicity of the resulting waste. Specially, oxidizing reagents containing transition metals such as KMnO4, K2Cr2O7, Pb(OAc)4 and others should be avoided.40 Among the oxidizing reagents that are environmentally benign, the most important ones are hydrogen peroxide, organic hydroperoxides and molecular oxygen. In the present Perspective and for the sake of conciseness, we will focus on those reports that have described the use of MOFs as oxidation catalysts for these three types of oxidants considered “green”. While hydrogen peroxide is almost perfect as oxidant due to high oxygen content and the formation of water as the only byproduct, the two main problems of this reagent are the lack of miscibility in organic solvents and its high cost. Organic hydroperoxides have the advantage of being soluble in most organic solvents and miscible with most organic substrates, but again the cost of these reagents is high.When employing hydroperoxides one point that should always be studied is the selectivity of the process with respect to these reagents. In other words, besides selectivity with respect to organic substrates, it is also necessary to know which percentage of hydroperoxides have been decomposed spuriously by the catalyst and which percentage have been successfully employed to transform the organic substrates. In general oxidant to substrates molar ratios as close as possible to the unity should be employed. Even more, frequently the substrate is cheaper than the hydroperoxides and, then, the oxidizing reagent can be employed in substoichiometric amounts to maximize its selectivity towards oxidation.
In terms of cost and availability, molecular oxygen is the ideal terminal oxidant but the challenge is how to direct its reactivity towards a single product. Oxidations using molecular oxygen are generally characterized by a considerable lack of selectivity and the formation of complex reaction mixtures that are frequently useless from the industrial point of view. One way to circumvent this lack of selectivity when using oxygen is to keep conversion low while in the case of use of hydroperoxides conversion and selectivity can be much higher and this can justify their use.
The reagents employed in asymmetric synthesis deserve one special comment. In this case, transmission of chirality to the products typically requires a bulk reagent that increases the free energy discrimination between the two transition states leading to each enantiomer. For this reason, N-oxides and iodosylbenzene are nucleophilic co-catalysts and oxidizing reagents, respectively, which are typically employed to enhance asymmetric induction.41
This Perspective has been organized by presenting examples of the use of MOFs as oxidation catalysts for a few types of organic compounds such as cycloalkanes and alkenes and alcohol oxidation. A final section deals with the use of MOFs as solid catalysts for enantioselective oxidations. The reader should consult the recent reviews covering the use of MOFs as catalysts for organic reactions for a comprehensive coverage of the use of MOFs as heterogeneous catalysts.1,16 Rather than being exhaustive, our purpose has been to describe the cutting edge of the current status of MOFs as oxidation catalysts using hydroperoxides/oxygen. We have constrained ourselves to examples that can be of large industrial or synthetic relevance such as oxidation of hydrocarbons and alcohols.
2. Oxidation of cycloalkanes
A copper-based MOF having a trinuclear triangle cluster namely [Cu3(μ3-OH)(μ-pz)3(EtCOO)2(H2O)] (Hpz = pyrazole) has been reported for peroxidative oxidation of cyclopentane and cyclohexane using hydrogen peroxide as oxidant in combination with nitric acid in acetonitrile/water medium under mild conditions.42 It has been shown that the presence of nitric acid is essential for the occurrence of the oxidation reaction in very high overall selectivity (presumably close to 100%) toward the formation of the corresponding alcohols and ketones. In all the conditions tested, cyclohexanol (25.1%) was the major product accompanied with a small amount of cyclohexanone (2.8%). No traces of typical byproducts for peroxidative oxidations of cycloalkanes namely diketones, diols, etc. were observed. Increasing the amount of catalyst and hydrogen peroxide resulted in increase of the percentage conversion. It is, however, remarkable that this copper MOF could be stable in the presence of nitric acid and water. Since, as commented earlier, MOF stability is a key issue in heterogeneous catalysis, convincing evidence of long-term stability should be presented.Recently, iron coordinated to 1,3,5-benzenetricarboxylate (BTC) [Fe(BTC)] metal–organic framework has been commercialized by BASF and is available from Sigma Aldrich under the name of Basolite F 300. Although the exact crystal structure of Fe(BTC) is still unknown, it is likely to possess a similar structure and environment as in MIL-10043 (Fe(III) coordinated with 1,3,5-benzenetricarboxylate), a highly crystalline MOF whose structure has been solved, but exhibits a different XRD than the commercial Basolite F 300. The aerobic oxidation of cyclooctane has been reported using N-hydroxyphthalimide loaded on Fe(BTC) [NHPI/Fe(BTC)] as a heterogeneous catalyst under solvent free conditions (Scheme 2).44 It has been demonstrated that a selectivity to cyclooctanol and cyclooctanone above 90% could be achieved at 30% conversion in 6 h. Increasing the reaction time further, decreases the selectivity towards cyclooctanol and cyclooctanone, a fact that is general in those reactions in which the primary reaction products can undergo secondary reactions. On the other hand, aerobic oxidation of tetralin showed the selectivity of tetranol and tetralone to 90% in 3 h with 33% conversion. This observed selectivity is quite comparable to other catalytic systems reported with peroxides as oxidant and supports the operation of a radical chain mechanism for the autooxidation reaction.
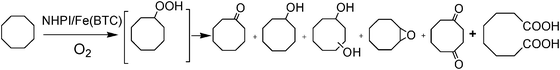 | ||
| Scheme 2 Aerobic oxidation of cyclooctane catalyzed by NHPI/Fe(BTC). | ||
The progress in this area should be achieved by showing the generality of this NHPI/Fe(BTC) system for the aerobic oxidation of other hydrocarbons including the industrially relevant cyclohexane and also acyclic alkanes. What appears reasonable is that considering that autooxidation using air is a general process, the target should be a better control of the selectivity at moderate conversions. MOFs as catalysts can be suitable materials to achieve this goal since confinement of radicals in a restricted reaction cavity can be a general strategy to control their reactivity. It is interesting to note that due to the diffusion limitations conventional large pore zeolites and particularly NaY were found to be not adequate to develop an analogous NHPI/Fe–NaY system. Also, Jacobs and co-workers reported an NHPI loaded on silica and observed that the activity and selectivity towards cyclohexane oxidation was low.45
3. Oxidation of benzylic compounds
Aerobic oxidation of alkyl aromatics and benzylic positions is a process of large industrial relevance that can be typically promoted by metal carboxylates in strong acid media.46 Since most MOFs are crystalline porous metal carboxylates there is a reasonable chance that they can also be active for this reaction, although the problem of stability against strong acids is going to pose a strong limitation to the use of MOFs. Due to the boiling point and easy analysis of the reaction products, tetralin (see the structure in Fig. 3) can be considered a suitable model compound for this important oxidation.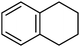 | ||
| Fig. 3 Structure of tetralin. | ||
MIL-101 (chromium terephthalate) has been used as catalyst for oxidation of tetralin in the presence of tert-butyl hydroperoxide (TBHP) as well as pivalaldehyde/O2 as oxidants.47 Under optimized conditions, oxidation of tetralin resulted in 70% conversion with 84.8% selectivity with TBHP (3 eq.), while in the presence of pivalaldehyde/O2 MIL-101 promotes higher tetralin conversion and selectivity reaching 93% and 86%, respectively. The nature of solvent and the removal of water were found to play a vital role. The catalyst showed very good stability under both reaction conditions and was reused for five runs without much loss in the catalytic activity using TBHP as oxidant.47
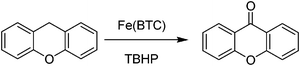
We were interested in exploring the catalytic activity of commercially available Fe(BTC) as an oxidation catalyst and hence oxidation of xanthene was tested with oxygen and hydrogen peroxide and resulted in no oxidation. In contrast, the combination of Fe(BTC) as catalyst and TBHP as oxidant leads to the transformation of xanthene to xanthone (Scheme 3).48 Under the optimized reaction conditions, more than 90% yield of xanthone was achieved with 1.5 equivalents of TBHP. Similarly, Cu3(BTC)2, a MOF that is also supplied by Sigma Aldrich as Basolite C 300, showed a slightly lower catalytic activity than Fe(BTC).48 The scope of these commercial MOFs was expanded to the oxidation of other benzylic compounds with high selectivity.
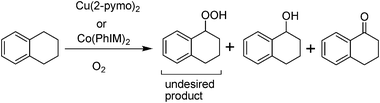
Aerobic oxidation of tetralin has been reported with Cu(2-pymo)2 (2-pymo = 2-hydroxypyrimidinolate) and Co(PhIM)2 (PhIM = benzoimidazolate) based MOFs as heterogeneous catalysts.49 When the aerobic oxidation was performed with Cu(2-pymo)2, 52% conversion of tetralin was observed accompanied by some undecomposed peroxides (Scheme 4). Co(PhIM)2 reached 23% tetralin conversion under the optimized conditions. The presence of hydroperoxides is highly detrimental for the quality of the reaction mixture of the oxidation of benzylic or cyclic alkanes due to the explosion risk associated with their presence in the reaction medium during the work up. In a way to increase the quality of the autooxidation mixture, minimizing the presence of hydroperoxides, combinations of Cu(2-pymo)2 and Co(PhIM)2 were used in a single catalytic system. It was hoped that in this way the high yields achieved using Cu(2-pymo)2 will be accompanied with a low hydroperoxide content due to the ability of Co(PhIM)2 to decompose them. As it was anticipated, the percentage conversion was found to be intermediate between that of a single Co(PhIM)2 and Cu(2-pymo)2 component accompanied with low amounts of the hydroperoxide.49 Both catalysts showed no leaching and could be reused without much loss in the catalytic activity.
In the previous examples, the constitutional metal ions present on the nodal framework positions are active as catalytic sites due to the presence of exchangeable coordination positions. But, besides providing active sites, MOFs can also be used as high surface area supports to include metal complexes that will act as the catalytic sites.
In these cases, MOFs act as supports to isolate other active sites incorporated inside the micropores increasing the catalytic activity by increasing dispersion and favoring accessibility. As an example of this strategy, Kegin type of polyoxometallate (POM) namely SiW12O404− has been incorporated in Cu2(4,4′-bpy)4(H2O)4 (bpy = bipyridine) and used as catalysts for oxidation of ethylbenzene using TBHP as oxidant. The copper MOF containing POM promoted ethylbenzene oxidation resulted in 56.8% conversion with 88% selectivity towards acetophenone at 70 °C, while the homogeneous POM showed only 27.5% conversion with 76% selectivity.50 The heterogeneous catalyst showed no leaching of copper and could be used three times without loss in the catalytic activity.
In general, when used as homogeneous catalysts for oxidation reactions in the liquid phase metallophthalocyanine (MPc) complexes51 exhibit limited reactivity due to aggregation through π-stacking and the formation of oligomers connected by oxo ligands. Analogously, tetrasulfophthalocyanine (FePcS) was reported as an efficient catalyst for the selective oxidation of aromatic compounds with TBHP.52 Heterogenization of FePcS over MIL-101 (FePcS/MIL-101) was reported as a heterogeneous catalyst for the oxidation of benzyl alcohol using TBHP as oxidant.53 The rate of oxidation was decreased when the loading of FePcS was increased from 5 to 15%. This negative effect of excessive guest loading is generally observed and can be attributed to the difficulty to access to the occluded FePcS when the loading is too high and diffusion inside the micropores is severely restricted. Although FePcS/MIL-101 showed better activity than unsupported FePcS, selectivity towards benzaldehyde was lower due to the formation of benzoic acid. However, one of the major problems of FePcS/MIL-101 is deactivation. Thus, FePcS/MIL-101 showed 35% conversion of benzyl alcohol in 15 min, but further increase of the time to 2 h showed the same conversion. This indicates that the catalyst has undergone deactivation during the course of reaction. This issue is, however, of capital importance in the field of MOF catalysts and efforts should have been devoted to analyze the spent catalyst in order to identify the poisons and the deactivation mechanism. In contrast to the case of benzyl alcohol, oxidation of 2,3,6-trimethylphenol was more facile with FePcS/MIL-101 than with FePcS due to the faster diffusion of aromatic compounds inside the framework.53
Hence, it is believed that loading of MPc onto MIL-101 would increase the catalytic activity of this hybrid material in terms of conversion and selectivity by isolating the active site and impeding aggregation and oligomerization. This higher stability of the active sites is reflected in an increase in the TON. For example, FePcF6@MIL-101 and RuPcF6@MIL-101 catalysts showed very high TON of 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 and 46300, respectively, for the aerobic oxidation of tetralin to 1-tetralone at 24 h while the corresponding unsupported FePcF6 showed only 6300. In contrast, (FePc-t-Bu4)2N supported on MIL-101 showed similar activity to (FePc-t-Bu4)2N due to the external localization of this large complex on the surface of MIL-101.54 This enhancement of catalytic activity of MPc on loading over MIL-101 shows the advantages of supporting and isolating active metal complexes in large area porous solids.
200 and 46300, respectively, for the aerobic oxidation of tetralin to 1-tetralone at 24 h while the corresponding unsupported FePcF6 showed only 6300. In contrast, (FePc-t-Bu4)2N supported on MIL-101 showed similar activity to (FePc-t-Bu4)2N due to the external localization of this large complex on the surface of MIL-101.54 This enhancement of catalytic activity of MPc on loading over MIL-101 shows the advantages of supporting and isolating active metal complexes in large area porous solids.
4. Oxidation of cycloalkenes
Oxidation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds constitutes important reactions in organic chemistry that can lead to a diverse array of derivatives. One of this type of reactions is epoxidation. Epoxides have a wide range of industrial applications as feedstocks for high added value products. Usually epoxidation is carried out in the presence of an oxidant such as hydrogen peroxide, TBHP or peracids. Although hydroperoxides in combination with suitable catalysts constitute an adequate system from the view point of green chemistry, due to their cost it would be desirable to develop alternative systems based on oxygen. Thus, in the present section we will discuss the various possibilities in which MOFs can act as heterogeneous catalysts in combination with one of the above oxidants, with a special emphasis on discussing the aerobic epoxidation. Also, it will be shown how selectivity is altered when the experimental conditions are changed.
C double bonds constitutes important reactions in organic chemistry that can lead to a diverse array of derivatives. One of this type of reactions is epoxidation. Epoxides have a wide range of industrial applications as feedstocks for high added value products. Usually epoxidation is carried out in the presence of an oxidant such as hydrogen peroxide, TBHP or peracids. Although hydroperoxides in combination with suitable catalysts constitute an adequate system from the view point of green chemistry, due to their cost it would be desirable to develop alternative systems based on oxygen. Thus, in the present section we will discuss the various possibilities in which MOFs can act as heterogeneous catalysts in combination with one of the above oxidants, with a special emphasis on discussing the aerobic epoxidation. Also, it will be shown how selectivity is altered when the experimental conditions are changed.
[Cu(H2btec)(bpy)]∞ (H4btec = 1,2,4,5-benzenetetracarboxylic acid and bpy = 2,2′-bipyridine) has been synthesised and used as catalyst for epoxidation of cyclohexene and oxidation of styrene by TBHP.55Oxidation of cyclohexene at 75 °C in 24 h resulted in 64.5% conversion with 1886 TON and 73.1% selectivity towards cyclohexene oxide (Scheme 5). This product was also accompanied with 7.7% and 19.2% of cyclohexanone and 2-cyclohexenone. On the other hand, oxidation of styrene at 75 °C resulted in 23.7% conversion with a TON of 673 in 24 h with 71% styrene oxide and 29% of benzaldehyde.55 It is interesting to note that TBHP was used in equimolar amounts and, therefore, it appears that the copper MOF exhibits a high selectivity for the oxidation from the point of view of the hydroperoxide consumption, minimizing spurious TBHP decomposition. However, the stability of catalyst in terms of leaching and reusability was not reported and this is an issue of large importance, particularly in oxidation reactions.
![Oxidation of cyclohexene with TBHP catalyzed by [Cu(H2btec)(bpy)]∞.](/image/article/2011/CY/c1cy00068c/c1cy00068c-s5.gif) | ||
| Scheme 5 Oxidation of cyclohexene with TBHP catalyzed by [Cu(H2btec)(bpy)]∞. | ||
It is interesting to note that epoxidation using Ti containing zeolites was a hot topic in heterogeneous catalysis in the 1990's.56–58 The outcome of this research is that Ti-Beta zeolite was a suitable solid catalyst for oxidation of alkenes using hydrogen peroxide as the reagent, but the selectivity towards epoxide was very low due to generally undesirable ring opening of epoxides.59 In order to increase selectivity towards epoxide absence of water and the use of hydrophobic catalysts with much larger pore sizes was needed. Thus, it appears that silanized Ti-MCM-41 in combination with TBHP is a good catalyst for epoxidation of propene and other alkenes.60 The Ti content of the catalysts has to be necessarily low in order to avoid the formation of TiO2 nanoparticles. Also, silanization is necessary to increase the stability of MCM-41 and expel water from the interior of MCM-41. One of the objectives of silanization was to increase the organophilicity and to reduce hydrophilicity of the MCM-41.61
In continuous effort to synthesise different MOFs, CoII4O(bdpb)3 (where H2bdpb 1,4-bis[(3,5-dimethyl)pyrazol-4-yl]benzene) was reported and its catalytic activity towards cyclohexene oxidation using TBHP as oxidant was studied.62 Negligible conversion of cyclohexene was observed in the absence of catalyst, while 27.5% conversion was achieved in its presence after 22 h. It has to be noted that the highest selectivity was towards the formation of peroxide and lowest towards cyclohexene epoxide (3%). Further decomposition of peroxide resulted in the formation of cyclohexenone. Powder XRD of the catalyst showed no sign for MOF decomposition and leaching experiments resulted in negligible amount of Co(II) ions in the hot filtration test. Although a 5% reduction in the conversion was observed from the first to the second run, further use of catalyst exhibited similar reactivity as the second run. This partial decrease of the catalyst activity was attributed to the pore blockage by organic compounds as evidenced by a decrease in BET surface area from 1485 to 1018 m2 g−1. Replacing TBHP with Ph3COOH dropped the catalytic activity significantly as the latter bulky hydroperoxide could not diffuse into the pores of MOF whereas TBHP is assumed to diffuse freely inside the framework.
Vanadium is one of the transition metals that exhibits general catalytic activity for oxidation reactions. Recently, vanadium containing metal–organic framework V-MIL-47 has been reported. MIL-47 consists of a porous terephthalate framework built from infinite chains of V4+O6 octahedra ordered in a three-dimensional orthorhombic structure that exhibits large pores. V-MIL-47 shows catalytic activity towards cyclohexene oxidation in the liquid phase using TBHP (2 eq.) (Scheme 6).63 The initial TOF exhibited by V-MIL-47 for cyclohexene conversion was 43 h−1 and increasing the time increased the percentage conversion to 50%. In the first 2 h, the formation of cyclohexene oxide reached to 15% and further time increase led to the formation of 1,2-cyclohexanediol, decreasing the epoxide selectivity to 5%. On the other hand, a significant amount of tert-butyl-2-cyclohexenyl-1-peroxide (30%) was also formed.63 It has been observed that the pores of V-MIL-47 were blocked by organic compounds and regeneration of catalyst was achieved by activating the catalyst at 523 K for 4 h.
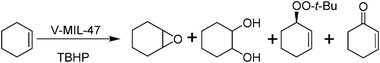 | ||
| Scheme 6 Oxidation of cyclohexene catalyzed by V-MIL-47 using TBHP as oxidant. | ||
Recently, MIL-101 has been reported as a heterogeneous catalyst for the allylic oxidation of alkenes using TBHP as oxidant.64 Although epoxidation of alkenes is a process of large industrial importance, allylic oxidation leading to the formation of α,β-enones or allylic alcohols is a very interesting reaction from the synthetic point of view. α,β-Enones can undergo Michael additions with a large variety of nucleophiles and are versatile synthetic intermediates. Under the optimized reaction conditions, cyclohexene showed 81% conversion with more than 93% selectivity towards 2-cyclohexenone. The temperature of the catalyst activation strongly influenced the yield of ketone. MIL-101 showed good stability as evidenced by the detection of less than 1 ppm of chromium by a hot filtration test. MIL-101 was reused for five times without much loss in the catalytic activity.
Although organic hydroperoxides are generally more suitable for the oxidation of organic compounds and cycloalkenes due to their solubility in organic solvents, hydrogen peroxide is a less costly oxidizing reagent. Hydrogen peroxide has as a general limitation the lack of solubility in common organic solvents, and thus generally the reactions employing hydrogen peroxide are conducted in two phases, a fact that complicates considerably diffusion and mass transfer between phases.
However, there are examples of the use of hydrogen peroxide in combination with MOFs. Thus, a porous coordination solid of nickel(II) dihydroxyterephthalate has been synthesized by the microwave-assisted method resulting in the formation of [Ni2(dhtp) (H2O)2]·8H2O (where dhtp = 2,5-dihydroxyterephthalate). [Ni2(dhtp) (H2O)2]·8H2O has been tested as catalyst for oxidation of cyclohexene using hydrogen peroxide as oxidant.65 At equal molar ratio of cyclohexenevs.hydrogen peroxide, 7.8% conversion was observed with 34.7% cyclohexene oxide, 21.7% cyclohexenone and 43.6% of 2-cyclohexenol. Increasing the molar ratio of cyclohexenevs.hydrogen peroxide to 3, 75.4% conversion was achieved, decreasing the selectivity of 2-cyclohexenol and the formation of 14.4% of 1,2-cyclohexanediol.65 The efficiency of utilization of hydrogen peroxide in the oxidation reaction has also been measured and determined that about 100% of the consumed hydrogen peroxide participates in cyclohexene oxidation. This fact is remarkable considering the tendency of hydrogen peroxide to undergo disproportionation to water and oxygen. The leaching experiment was performed with 3 eq. of hydrogen peroxide and the catalyst showed leaching (the percentage not mentioned) at 65 °C while at room temperature no leaching of nickel was observed.
Besides exploiting the catalytic activity of constitutional metals present in MOFs, these porous materials can also be used to include in the intracrystalline space some guests that can be the catalytic sites for the oxidation. Following a similar work as the one reported previously for ethylbenzene oxidation, polyoxotungstates [PW4O24]3− (PW4) and [PW12O40]3− (PW12) have been inserted electrostatically into the nanocages of the metal–organic framework MIL-101 at a loading between 5 and 14 wt% and used as catalyst for cyclohexene oxidation.66 With 2 equiv. of H2O2, 5% PW4/MIL-101 showed 76% substrate conversion after 3 h with 74% selectivity of cyclohexene oxide. Very similar results in terms of conversion and cyclohexene oxide selectivity were achieved for 5% PW12/MIL-101. The allylic oxidation products, 2-cyclohexene-1-ol and 2-cyclohexene-1-one (totally ca. 10%) were identified as the main byproducts, while the product of epoxide ring-opening, 1,2-trans-cyclohexane diol, was not found in the reaction mixture. This contrasts with the homogeneously catalyzed oxidation in the presence of PW4 or PW12 where the diol was the main byproduct. Elemental analysis and hot filtration test showed the absence of any tungsten (from the polyoxotungstates) or chromium (from MIL-101) traces in the liquid phase and that the reaction stops when the solid is removed. PW4/MIL-101 showed a slight decrease in the epoxide yield after three runs and this could be attributed to the decrease in the specific surface area and pore volume. Powder XRD shows amorphization of the MIL-101 catalyst due to partial decomposition during the cyclohexene oxidation with hydrogen peroxide.66
In continuous effort in synthesizing new porous MOFs, Hupp and co-workers reported the synthesis of a porous organic polymer67 (POP) containing free-base porphyrin subunits by the condensation of a bis(phthalic acid)porphyrin with tetra(4-aminophenyl)methane. Metallation by post-synthesis modification affords microporous materials incorporating either Fe or Mn(porphyrins) that have been shown to be active catalysts for styrene epoxidation using iodosylbenzene as oxidant.67 Metallated meso-tetrapentafluorophenylporphyrin (TPFPP) as homogenous complexes with Mn and Fe rapidly produce styrene epoxides, but with facile deactivation and degradation of the catalyst reaching a maximum TON of 780 in the case of (TPFPP)Mn, and 170 in the case of (TPFPP)Fe. In contrast, both the Fe- and Mn-PPOP show greater stability than the analogous homogenous catalysts and Mn-PPOP is active for more than 600 turnovers and Fe-PPOP with 300 without displaying any sign of decomposition. Although both catalysts were reused for three cycles, the activity was decreased in the second and third cycles. The loss of catalytic activity can be attributed to the oxidation of individual pyrrolic rings and not to the destruction of the polymer network as evidenced by SEM analysis of the spent catalyst. Demetallation of the material was also ruled out as ICP analysis showed the same metal content before and after catalytic oxidation.
In the previous examples, the oxidizing reagents have been organic hydroperoxides and occasionally hydrogen peroxide and iodoso compounds. Hydroperoxides are environmentally benign but it would be more desirable to use directly molecular oxygen as oxidant. The control of aerobic oxidations harnessing the reactivity to the desired products is a challenge in catalysis. In this context, [Cu(bpy)(H2O)2(BF4)2(bpy)] (bpy: 4,4′-bipyridine) has been reported as a heterogeneous catalyst for allylic oxidation of cyclohexene in the presence of oxygen as oxidant.68 In all the cases, the selectivity of 2-cyclohexenylhydroperoxide was more than 85% with only detectable amount of cyclohexene epoxide, 2-cyclohexenone and 2-cyclohexenol.
As it is commented earlier, the use of molecular oxygen as a terminal oxidant is always desirable and in this connection, a manganese(II) acetylacetonate complex has been immobilized to the amino functionalized isoreticular metal–organic framework IRMOF-3 through a one-step post-synthetic route providing heterogeneous catalyst IRMOF-3(Mn).69 This catalyst has been tested for the epoxidation of cyclohexene, cyclooctene and styrene using pivalaldehyde and oxygen as oxidant in toluene medium (Scheme 7). Cyclohexene showed 68% of conversion towards 92% of cyclohexene oxide accompanied with 8% of allylic products. Cyclooctene reached 60% conversion exhibiting 96% epoxide selectivity. Analogously, styrene showed 52% conversion with 80% selectivity towards styrene oxide. IRMOF-3(Mn) was reused for three cycles with a similar reactivity and selectivity in all the three substrates. Powder XRD showed no sign of decomposition of the Mn complex in the spent catalyst after the catalytic oxidation reaction and no leaching of Mn was observed. The major concern is the need of 2 equivalents of pivalaldehyde and no mechanism for the high selectivity observed was proposed.
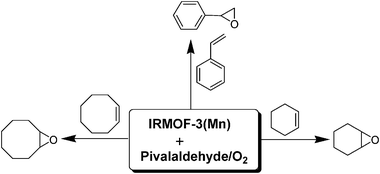 | ||
| Scheme 7 Epoxidation of olefins catalyzed by IRMOF-3(Mn) with pivalaldehyde/O2 as oxidant. | ||
After having observed a high catalytic activity for cyclooctane and benzyl amine oxidation, we were interested to make use of NHPI/Fe(BTC) as catalyst for cycloalkene oxidation. In this context, epoxidation of cyclooctene was performed using NHPI/Fe(BTC) as catalyst under neat conditions at 100 °C.70 This catalyst showed 9% conversion with 95% selectivity towards epoxide. An effort to increase the conversion to 48% reduced the selectivity of epoxide to 85%. Even though, this selectivity is still remarkable at such high conversion in the absence of solvent. Also, it has been demonstrated that the ring size plays an important role in determining the conversion and selectivity of the products. Thus, cyclopentene and cyclohexene undergo preferentially allylic oxidation under the same conditions in which epoxidation occurs for cyclooctene. The catalyst could be reused two times and its structural integrity was determined from powder XRD by comparing fresh and spent catalysts.
In contrast to the current state of the use of MOFs for oxidation of alkenes using oxygen, there is a lack in the literature reporting the use of zeolites for aerobic oxidation of alkenes. This is due to the fact that aluminosilicates are inert as oxidation catalyst and the presence of suitable transition metals is needed. However, as we have commented, transition metals in zeolites tend to evolve from framework to extra framework positions, rendering unstable materials whose catalytic activity vary depending on the state of the catalytic sites.
5. Oxidation of alcohols
Selective oxidation of primary alcohols to aldehydes is a highly relevant transformation in organic chemistry due to the properties and chemical reactivity of these carbonylic compounds that make aldehyde appropriate starting materials in many synthetic methods. In this connection, developing a suitable catalyst for this oxidation is a long standing research area. [Pd(2-pymo)2]n (2-pymo = 2-hydroxypyrimidinolate)71 has been synthesised with 32% wt of Pd and used for the selective aerobic oxidation of cinnamyl alcohol reaching 74% of selectivity towards the desired cinnamaldehyde in 20 h. Besides the applications and uses of cinnamaldehydes, oxidation of cinnamyl alcohol is considered a model reaction since there is an array of competitive pathways that can take place simultaneously to the alcohol oxidation including epoxidations, CC isomerization and even polymerization and hydrogenation. The hot filtration test did not show the presence of Pd and hence stability of the catalyst was assumed. However, since palladium in the homogeneous phase can be highly active even in very low concentrations and trace amounts,72,73 the possibility that Pd(2-pymo)2 is acting as a solid reservoir providing some palladium to the solution was not conclusively ruled out. It has also been shown that the Pd(2-pymo)2 catalyst is active for Suzuki cross coupling and for the shape selective hydrogenation of olefins.71
[Zn4O(btb)2]8 (H3btb = 1,3,5-benzenetribenzoic acid), MOF-177 has been used as support for loading of Pt nanoparticles in the range of 2–5 nm and as catalyst for hydrogen storage and aerobic oxidation of benzylic and allylic alcohols at room temperature under base and solvent free conditions.74 Benzylic substrates showed very good reactivity with TON > 500 (Scheme 8) and cinnamyl alcohol with 385. In contrast, aerobic oxidation of iodo and bromo substituted benzyl alcohols failed due to their steric hindrance that impedes these substrates to reach the active sites. Aliphatic alcohols were found to be unreactive under identical conditions. After using MOF-177 as catalyst for the aerobic oxidation of benzyl alcohol, powder XRD of the used material revealed that breakdown of the MOF host lattice occurred leading to a negligible conversion of benzyl alcohol in an attempted reuse. This lack of MOF-177 stability could be anticipated from the fact that the Zn4O cluster is highly water sensitive and water is produced as a byproduct during the oxidation.
![Aerobic oxidation of benzyl alcohol to benzaldehyde using Pt nanoparticles supported on [Zn4O(bdc)2]8.](/image/article/2011/CY/c1cy00068c/c1cy00068c-s8.gif) | ||
| Scheme 8 Aerobic oxidation of benzyl alcohol to benzaldehyde using Pt nanoparticles supported on [Zn4O(bdc)2]8. | ||
Recently, Haruta and co-workers deposited Au clusters on several MOFs such as MOF-5, [Zn4O(bdc)3]n (bdc = benzene-1,4-dicarboxylate), Al-MIL-53, [Al(OH)(bdc)]n and Cu3(BTC)2. A volatile organogold complex, Me2Au(acac) (acac = acetylacetonate), and MOFs were ground in an agate mortar in air for 20 min at room temperature. Then, the mixture was treated in a N2 stream having 10 vol% H2 at 120 °C for 2 h to obtain Au supported on different MOFs. The very simple grinding procedure for gold incorporation is remarkable. These gold containing catalysts were tested for the liquid phase aerobic oxidation of benzyl alcohol and 1-phenylethanol in methanol.75Au/MOF-5 exhibited 82% conversion of benzyl alcohol in the presence of a base resulting in the formation of 66% of methyl benzoate and 3% benzaldehyde while in the absence of a base, 69% conversion with 23% methyl benzoate and 31% benzaldehyde. Au/Al-MIL-53 showed 98% conversion of benzyl alcohol with the selectivity of 2% of benzaldehyde and 77% of methyl benzoate.75Gold nanoparticles supported on MOF-5 (0.5 wt % Au with a mean diameter of 4.8 ± 2.2 nm) exhibited the highest catalytic activity among all the Au supported on different MOFs by solid grinding for 1-phenylethanol oxidation in the presence of a base affording 79% yield of acetophenone.75
Recently, nanoparticles of gold have been supported also on MIL-101 using different methods and Au deposited by colloidal deposition with PVP as a protecting agent was the sample exhibiting the highest catalytic activity for aerobic oxidation of alcohols to aldehydes under base free conditions.76 This may be due to lower mean diameter of gold nanoparticles (2.3 nm) compared to other methods of loading on MIL-101. However, how diffusion of gold nanoparticles inside MOF occurs is a matter that deserves further study. Benzylic alcohols were oxidized to their corresponding aldehydes in high conversion (>95%) and selectivity (>99%).76 Secondary aliphatic alcohols showed higher activity than primary counterparts (Scheme 9). Alcohols with heteroatoms were also smoothly oxidized with high conversion and selectivity. The catalyst showed no leaching of gold and TEM analysis confirmed that the particle size of gold before and after oxidation reaction remains without aggregation. The catalyst showed consistent yield (>99%) for the oxidation of 4-methoxybenzyl alcohol until the sixth cycle. This catalytic system showed very high TOF for the aerobic oxidation of 1-phenylethanol with 29300 h−1.76 This high catalytic activity is probably due to the high dispersion of Au nanoparticles in combination with a beneficial synergetic effect of the MIL-101 support. As is well-known, the support may play an important role, either direct or indirect, in determining the activity and selectivity of gold.77 In addition, the higher catalytic activity of this gold-containing MOF could be explained from the fact of good chemical resistance to water and organic solvents by MIL-101 and the larger pore size of MIL-101 (ca. 30 Å), facilitating mass transfer of reactant/product molecules inside the pores.
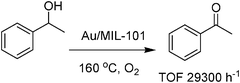 | ||
| Scheme 9 Aerobic oxidation of 1-phenylethanol catalyzed by Au/MIL-101. | ||
Aerobic oxidation of benzyl alcohols has been reported using Cu3(BTC)2 as catalyst in the presence of a base and catalytic amount of TEMPO in acetonitrile medium (Scheme 10).78 Although this catalyst showed moderate catalytic activity for benzyl alcohols, they exhibited low activity towards the oxidation of aliphatic, secondary and cyclic alcohols. In addition to the lack of general scope of Cu3(BTC)2 in combination with TEMPO, a leaching test showed 1 ppm of copper and the catalyst undergoes some changes in its crystal structure as evidenced by XRD. It has been demonstrated that benzoic acid formed during the reaction could lead to deactivation of catalyst by coordination with the free metal centres. Reactivation is certainly an interesting issue. Considering that the structure of common MOFs is based on metal–carboxylate interactions it may happen that carboxylic acids are general poisons for any reaction requiring free coordination sites of framework nodal metals by strong interaction with the metal sites. Since carboxylic acids can generally be formed at least in minor amounts in the aerobic oxidation of primary alcohols, it is important to show if the poisoning by the presence of carboxylic acids is a general deactivation mechanism.
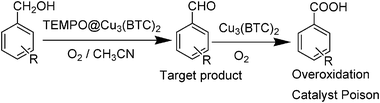 | ||
| Scheme 10 Deactivation of Cu3(BTC)2 by over oxidation of benzaldehyde. | ||
6. Enantioselective oxidation by chiral MOFs
MOFs are constituted by nodal metallic ions or clusters connected by rigid linkers. As we have commented in comparison with zeolites, the organic components allow large flexibility in design and modification of the linker. One special case is when the linker exhibits chirality and only one of the enantiomers is used in the preparation of MOFs. Depending on the synthesis conditions homochiral MOFs are obtained that can be suitable as solid catalysts to transmit chirality starting from an achiral substrate (asymmetric induction by catalysis).33 The field of chiral MOFs has been recently reviewed and among the chiral linkers that have been reported so far the most widely used are those based on 1,1′-binaphthyls having substituents in the periphery to coordinate with metallic nodes. Also, one remarkable example in which a chiral metal salen complex has been used as a linker has been reported by the group of Hupp.79Scheme 11 shows some of the chiral linkers that have been employed for the synthesis of chiral MOFs.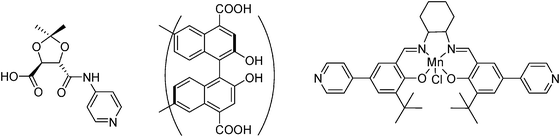 | ||
| Scheme 11 Chiral linkers used for the synthesis of chiral MOFs. | ||
Besides the use of chiral linkers, another possible strategy to develop asymmetric MOF catalysts could be to start with a non-chiral MOF and subsequently modify it with chiral spectators accommodated inside the MOF cavities.
It is interesting to compare this situation with zeolites and mesoporous silicas. The synthesis of homochiral zeolites has been a target in the 1990s and even now and not much success has been achieved.80Zeolites require the use of an organic template and by using chiral organic templates, it was expected to obtain homochiral zeolites. However, the harsh conditions (high temperature, strongly basic media, long reaction times etc.) required for the synthesis of zeolites has resulted in poor results. Since chiral zeolites are difficult to obtain, the possibility to include chiral spectators inside zeolite cavities has been more frequently studied but also the enantiomeric excesses obtained have been very low.81
Much better results have been obtained starting with mesoporous silica or organosilicas and introducing in the inorganic framework chiral organic components or metallic complexes to produce chiral solids.82,83 Particularly successful has been the development of chiral periodic mesoporous organosilicas (ChiMO).84 This strategy however has the disadvantage of dedicated organic synthesis to obtain chiral silanes needed for the hybrid organosilica and subsequent use of organic surfactant (that later has to be removed) in the synthesis of the mesoporous solids. In recent times, chiral MOFs have been successfully reported as catalysts for many enantioselective reactions including epoxidation of alkenes.85–87
Asymmetric inductions typically are carried out at low temperatures and under mild conditions.88 In fact, it is very frequently observed that ambient temperature or the presence of acids and bases spoils enantiomeric excess. These mild conditions are very well suited to preserve the crystal structure of MOFs and in this regard reaction conditions are compatible with MOF stability. The key point is again diffusion of reagents and products through the pores in the liquid phase.
Concerning enantioselective oxidations, one key point is the reagents that are most commonly employed. Typically the reagent should transfer oxygen to the active catalytic site in such a way that only one enantiomer of the active site is formed. This generally requires the use of bulky reagents. Thus, although hypochlorite and hydroperoxides have sometimes been used for oxidations other reagents such as N-oxides and iodoso compounds are more often used.
The selection of this type of bulky oxidant such as iodosylbenzene for enantioselective oxidation is not very fortunate from the “green point of view” since this is a stoichiometric reagent that would generate a large amount of waste. However, due to the influence of steric hindrance in the asymmetric induction, it is almost impossible to induce chirality using molecular oxygen as the direct oxidant. Nevertheless, a catalytic cycle in which under stoichiometric amounts of iodosylbenzene are used and its reoxidation is performed by oxygen could be feasible.
Besides reagents another point of interest is the type of products that would be of industrial interest. In this sense, chiral epoxides are one of the preferred targets since the reactivity of these compounds allows later to transmit chirality to a large variety of mono and difunctional compounds.89 It is worth commenting that chiral epoxidations of allylic alcohols reported by Sharpless using homogeneous chiral titanium complexes as catalysts constituted a milestone in this area.90 As an example of how chiral MOFs can be used as catalysts for alkene epoxidation, Nguyen and co-workers studied the enantioselective epoxidation of chromenes. In this example, an Mn(III)–Schiff base complex forming part of the MOF linker in Zn2(bpdc)2L·10DMF·8H2O91 (L = (R,R)-(−)-1,2-cyclohexanediamino-N,N′-bis(3-tert-butyl-5-(4-pyridyl)salicylidene)MnIIICl and bpdc = biphenyldicarboxylate) was used as a solid catalyst (Scheme 12).91 Although, L as a homogeneous catalyst showed higher initial activity than Zn2(bpdc)2L·10DMF·8H2O, at long reaction times the latter solid showed better activity by exhibiting higher turnover number (TON) of 1430 with 82% ee while L showed 88% ee with TON of around 600. Under these experimental conditions, leaching of Mn was determined to occur between 4 and 7% in each cycle. This loss of Mn has been proposed to be responsible for the decrease of yield and TON observed for Zn2(bpdc)2L·10DMF·8H2O upon reuse. The gradual decrease in catalytic activity is not accompanied by a decrease in ee.91
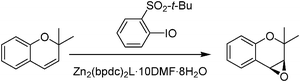 | ||
| Scheme 12 Enantioselective epoxidation of 2,2-dimethyl-2H-chromene by 2-tert-butylsulfonyliodobenzene as oxidant using Zn2(bpdc)2L·10DMF·8H2O as a heterogeneous catalyst. | ||
The reader is referred to recent reviews33 on chiral MOFs for a more complete coverage on this topic but for the present purpose, we should stress that a methodology in which molecular oxygen or organic hydroperoxides are the final oxidants and some chiral cocatalysts are used in small percentages is still to be developed, which will be a significant development in the field of catalytic enantioselective oxidations.
7. Conclusions and future perspectives
From the above examples on the use of MOFs as heterogeneous catalysts for oxidation reactions it can be concluded that these materials are going to be widely used for liquid phase reactions expanding considerably the range of organic compounds that will be selectively oxidized. All these works should demonstrate conclusively the stability of the framework and if possible they should address the deactivation mechanism. General statements like observing the same catalytic behaviour for a few additional cycles are not sufficient. Temporal profiles after several uses should be provided showing not only conversion data at a given time but also initial reaction rates. In addition long term stability would be better checked by performing continuous flow experiments rather than batch reactions. One figure of merit that would be interesting to know is maximum productivity before the MOFs become deactivated and the reactivation procedures. Also, it would be of large interest, for those cases in which real industrial applications could be envisioned, to determine which are the products derived from the organic linker under the reaction conditions even if this linker oxidation requires very long time and harsh conditions.Concerning the oxidizing reagents and even though organic hydroperoxides are simpler to be used due to solubility, more efforts should be devoted to carry out oxidations with hydrogen peroxide. This certainly requires the use of MOFs that are stable in the presence of water, a fact that is still not general in many cases. Also, it is easily anticipated that oxidation using molecular oxygen in combination with MOFs as catalysts is a field that will develop intensively in the near future. In this area besides the use of nodal metals a strategy in which micropores contain metal nanoparticles such as gold or palladium seems very promising. Also, the combination of MOFs with organic promoters of radical chain mechanisms has large potential in aerobic oxidations.
Concerning the substrates, it would be highly relevant to apply MOFs for oxidation of cyclohexane, epoxidation of propene and oxidation of linear alcohols in non-aqueous medium. In these three targets, there is no doubt that MOFs, sometimes following the lead of previous work with zeolites, are very promising as solid catalysts.
Finally considering the easy design and simplicity of the synthesis of chiral MOFs it can be foreseen that in the near future, this field will grow enormously, also for oxidation reactions and some excellent chiral solid catalysts will be developed in the near future.
Acknowledgements
Financial support by the Spanish DGI (CTQ2009-11587 and CTQ2010-18671 and Consolider MULTICAT) is gratefully acknowledged. Funding of European Commission through an integrated project (MACADEMIA) is also acknowledged.References
- Z. Wang, G. Chen and K. Ding, Chem. Rev., 2009, 109, 322 CrossRef CAS.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- A. Corma, H. Garcia and F. X. Llabres i Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- M. Kondo, T. Okubo, A. Asami, S. Noro, T. Yoshitomi, S. Kitagawa, T. Ishii, H. Matsuzaka and K. Seki, Angew. Chem., Int. Ed., 1999, 38, 140–143 CrossRef CAS.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 25, 8876–8883.
- J. L. C. Rowsell, A. R. Millward, K. S. Park and O. M. Yaghi, J. Am. Chem. Soc., 2004, 126, 5666–5667 CrossRef CAS.
- M. Eddaoudi, H. Li and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 1391–1397 CrossRef CAS.
- R. Kitaura, K. Seki, G. Akiyama and S. Kitagawa, Angew. Chem., Int. Ed., 2003, 42, 428–431 CrossRef CAS.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O’Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS.
- L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- G. Ferey, Nature, 2005, 436, 187–188 CrossRef CAS.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem.–Eur. J., 2010, 16, 8530 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Adv. Synth. Catal., 2010, 352, 711 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Adv. Synth. Catal., 2009, 351, 2271 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2010, 46, 6476–6478 RSC.
- F. Gándara, B. Gomez-Lor, E. Gutiérrez-Puebla, M. Iglesias, M. A. Monge, D. M. Proserpio and N. Snejko, Chem. Mater., 2008, 20, 72–76 CrossRef CAS.
- K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81 CrossRef CAS.
- L. Alaerts, E. Seguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. De Vos, Chem.–Eur. J., 2006, 12, 7353 CrossRef CAS.
- B. Gomez-Lor, E. Gutierrez-Puebla, M. Iglesias, M. A. Monge, C. Ruiz-Valero and N. Snejko, Inorg. Chem., 2002, 41, 2429 CrossRef CAS.
- K. S. Suslick, P. Bhyrappa, J. H. Chou, M. E. Kosal, S. Nakagaki, D. W. Smithenry and S. R. Wilson, Acc. Chem. Res., 2005, 38, 283 CrossRef CAS.
- C. N. Kato, M. Hasegawa, T. Sato, A. Yoshizawa, T. Inoue and W. Mori, J. Catal., 2005, 230, 226 CrossRef CAS.
- M. Ranocchiari and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2011, 13, 6388–6396 RSC.
- F. Vermoortele, R. Ameloot, A. Vimont, C. Serre and D. De Vos, Chem. Commun., 2011, 47, 1521–1523 RSC.
- A. Corma, in Recent Advances in the Science and Technology of Zeolites and Related Materials, Pts A–C, ed. E. VanSteen, M. Claeys and L. H. Callanan, 2004, vol. 154, pp. 25–40 Search PubMed.
- J. L. Sun, C. Bonneau, A. Cantin, A. Corma, M. J. Diaz-Cabanas, M. Moliner, D. L. Zhang, M. R. Li and X. D. Zou, Nature, 2009, 458, 1154–U1190 CrossRef CAS.
- A. Philippaerts, S. Paulussen, A. Breesch, S. Turner, O. I. Lebedev, G. V. Tendeloo, B. Sels and P. A. Jacobs, Angew. Chem., Int. Ed., 2011, 50, 3947–3949 Search PubMed.
- T. Tatsumi, H. Sakashita and K. Asano, J. Chem. Soc., Chem. Commun., 1993, 1264–1265 RSC.
- W. Lin, Top. Catal., 2010, 53, 869–875 CrossRef CAS.
- Y. Gao and K. B. Sharpless, J. Org. Chem., 1988, 53, 4081–4084 CrossRef CAS.
- C. W. Jones, Applications of Hydrogen Peroxide and Derivatives, Royal Society of Chemistry, Thomas Graham House, London, 1999 Search PubMed.
- A. Corma and H. Garcia, Chem. Rev., 2002, 102, 3837–3892 CrossRef CAS.
- G. Bellussi, A. Carati, M. G. Clerici, A. Esposito, R. Millini and F. Buonomo, Belg. Pat. 1001038, 1989 Search PubMed.
- M. Taramasso, G. Perego and B. Notari, US 4410501, 1983 Search PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press: New York, 1998, p. 30 Search PubMed.
- A. Fatiadi, Synthesis, 1987, 85–127 CrossRef CAS.
- E. N. Jacobsen, in Catalytic Asymmetric Synthesis, ed. I. Ojima, VCH, New York, 1993, pp. 159–202 Search PubMed.
- C. Di Nicola, Y. Yu. Karabach, A. M. Kirillov, M. Monari, L. Pandolfo, C. Pettinari and A. J. L. Pombeiro, Inorg. Chem., 2007, 46, 221–230 CrossRef.
- P. Horcajada, S. Surble, C. Serre, D. Y. Hong, Y. K. Seo, J. S. Chang, J. M. Greneche, I. Margiolaki and G. Ferey, Chem. Commun., 2007, 2820–2822 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem.–Eur. J., 2011 DOI:10.1002/chem.201002664.
- I. Hermans, J. Van Deun, K. Houthoofd, J. Peeters and P. A. Jacobs, J. Catal., 2007, 251, 204–212 CrossRef CAS.
- K. Weissermel and H.-J. Harpe, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 4th edn, 2003 Search PubMed.
- J. Kim, S. Bhattacharjee, K.-E. Jeong, S.-Y. Jeong and W.-S. Ahn, Chem. Commun., 2009, 3904–3906 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, J. Catal., 2009, 267, 1–4 CrossRef CAS.
- F. X. Llabrés i Xamena, O. Casanova, R. Galiasso Tailleur, H. Garcia and A. Corma, J. Catal., 2008, 255, 220–227 CrossRef CAS.
- F. Yu, P.-Q. Zheng, Y.-X. Long, Y.-P. Ren, X.-J. Kong, L.-S. Long, Y.-Z. Yuan, R.-B. Huang and L.-S. Zheng, Eur. J. Inorg. Chem., 2010, 4526–4531 CrossRef CAS.
- A. B. Sorokin, J.-L. Seris and B. Meunier, Science, 1995, 268, 1163 CrossRef CAS.
- O. V. Zalomaeva, A. B. Sorokin and O. A. Kholdeeva, Green Chem., 2010, 12, 1076–1082 RSC.
- O. V. Zalomaeva, K. A. Kovalenko, Y. A. Chesalov, M. S. Mel'gunov, V. I. Zaikovskii, V. V. Kaichev, A. B. Sorokin, O. A. Kholdeeva and V. P. Fedin, Dalton Trans., 2011, 40, 1441–1444 RSC.
- E. Kockrick, T. Lescouet, E. V. Kudrik, A. B. Sorokin and D. Farrusseng, Chem. Commun., 2011, 47, 1562–1564 RSC.
- K. Brown, S. Zolezzi, P. Aguirre, D. Venegas-Yazigi, V. Paredes-Garcia, R. Baggio, M. A. Novak and E. Spodine, Dalton Trans., 2009, 1422–1427 RSC.
- A. Corma, M. T. Navarro and J. Perez-Pariente, J. Chem. Soc., Chem. Commun., 1994, 147 RSC.
- A. Corma, U. Diaz, M. E. Domine and V. Fornes, Chem. Commun., 2000, 137–138 RSC.
- A. Corma and H. García, Chem. Rev., 2002, 102, 3837–3892 CrossRef CAS.
- A. Corma, P. Esteve, A. Martinez and S. Valencia, J. Catal., 1995, 152, 18–24 CrossRef CAS.
- A. Corma, M. E. Domine, J. A. Gaona, J. L. Jorda, M. T. Navarro, F. Rey, J. Perez-Pariente, J. Tsuji, B. McCulloch and L. T. Nemeth, Chem. Commun., 1998, 2211 RSC.
- A. Corma, J. L. Jorda, M. T. Navarro and F. Rey, Chem. Commun., 1998, 1899–1900 RSC.
- M. Tonigold, Y. Lu, B. Bredenkotter, B. Rieger, S. Bahnmuller, J. Hitzbleck, G. Langstein and D. Volkmer, Angew. Chem., Int. Ed., 2009, 48, 7546–7550 CrossRef CAS.
- K. Leus, I. Muylaert, M. Vandichel, G. B. Marin, M. Waroquier, V. V. Speybroeck and P. V. Der Voort, Chem. Commun., 2010, 46, 5085–5087 RSC.
- N. V. Maksimchuk, K. A. Kovalenko, V. P. Fedin and O. A. Kholdeeva, Adv. Synth. Catal., 2010, 352, 2943–2948 Search PubMed.
- J. S. Lee, S. B. Halligudi, N. H. Jang, D. W. Hwang, J.-S. Chang and Y. K. Hwang, Bull. Korean Chem. Soc., 2010, 31, 1489–1495 Search PubMed.
- N. V. Maksimchuk, K. A. Kovalenko, S. S. Arzumanov, Y. A. Chesalov, M. S. Melgunov, A. G. Stepanov, V. P. Fedin and O. A. Kholdeeva, Inorg. Chem., 2010, 49, 2920–2930 CrossRef CAS.
- A. M. Shultz, O. K. Farha, J. T. Hupp and S. T. Nguyen, Chem. Sci., 2011, 2, 686–689 RSC.
- D. Jiang, T. Mallat, D. M. Meier, A. Urakawa and A. Baiker, J. Catal., 2010, 270, 26–33 CrossRef CAS.
- S. Bhattacharjee, D.-A. Yang and W.-S. Ahn, Chem. Commun., 2011, 47, 3637–3639 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Angew. Chem., Int. Ed., 2011 Search PubMed , in reviewing process.
- F. X. Llabrés i Xamena, A. Abad, A. Corma and H. Garcia, J. Catal., 2007, 250, 294–298 CrossRef.
- A. H. M. de Vries, J. Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285–3288 CrossRef CAS.
- M. T. Reetz and J. G. de Vries, Chem. Commun., 2004, 1559–1563 RSC.
- S. Proch, J. Herrmannsdorfer, R. Kempe, C. Kern, A. Jess, L. Seyfarth and J. Senker, Chem.–Eur. J., 2008, 14, 8204–8212 CrossRef CAS.
- T. Ishida, M. Nagaoka, T. Akita and M. Haruta, Chem.–Eur. J., 2008, 14, 8456–8460 CrossRef CAS.
- H. Liu, Y. Liu, Y. Li, Z. Tang and H. Jiang, J. Phys. Chem. C, 2010, 114, 13362–13369 CrossRef CAS.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ACS Catal., 2011, 1, 48–53 Search PubMed.
- S.-H. Cho, T. Gadzikwa, M. Afshari, S. T. Nguyen and J. T. Hupp, Eur. J. Inorg. Chem., 2007, 4863–4867 CrossRef CAS.
- J. H. Yu and R. R. Xu, J. Mater. Chem., 2008, 18, 4021–4030 RSC.
- M. Benitez, G. Bringmann, M. Dreyer, H. Garcia, H. Ihmels, M. Waidelich and K. Wissel, J. Org. Chem., 2005, 70, 2315–2321 CrossRef CAS.
- C. Baleizão and H. Garcia, Chem. Rev., 2006, 106, 3987–4043 CrossRef CAS.
- M. Alvaro, M. Benitez, D. Das, B. Ferrer and H. Garcia, Chem. Mater., 2004, 16, 2222–2228 CrossRef CAS.
- C. Baleizão, B. Gigante, D. Das, M. Alvaro, H. Garcia and A. Corma, Chem. Commun., 2003, 1860–1861 RSC.
- M. Banerjee, S. Das, M. Yoon, H. J. Choi, M. H. Hyun, S. M. Park, G. Seo and K. Kim, J. Am. Chem. Soc., 2009, 131, 7524–7525 CrossRef CAS.
- F. Song, C. Wang, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132, 15390–15398 CrossRef.
- K. S. Jeong, Y. B. Go, S. M. Shin, S. J. Lee, J. Kim, O. M. Yaghi and N. Jeong, Chem. Sci., 2011, 2, 877–882 RSC.
- C. Baleizão, B. Gigante, H. Garcia and A. Corma, J. Catal., 2003, 215, 199–207 CrossRef CAS.
- Y. Gao, J. M. Klunder, R. M. Hanson, H. Masamune, S. Y. Ko and K. B. Sharpless, J. Am. Chem. Soc., 1987, 109, 5765–5780 CrossRef CAS.
- T. Katsuki and V. Martin, Org. React., 1996, 48, 1–299 CAS.
- S. H. Cho, B. Ma, S. T. Nguyen, J. T. Hupp and T. E. Albrecht-Schmitt, Chem. Commun., 2006, 2563–2565 RSC.
| This journal is © The Royal Society of Chemistry 2011 |