Adjusting framework ionicity to favour crystallisation of zeolites with strained structural units. Periodic quantum chemical studies
Evangelina
Martínez-Morales
and
Claudio M.
Zicovich-Wilson
*
Facultad de Ciencias, Universidad Autónoma del Estado de Morelos, Av Universidad 1001, Col Chamilpa, 62209, Cuernavaca (MOR), Mexico. E-mail: claudio@uaem.mx; Fax: +52 777 329 7040; Tel: +52 777 329 7020
First published on 6th June 2011
Abstract
Porosity in silicates is reached against the thermodynamic trend that favours the formation of the most dense phases. Porous silica materials, like Si-zeolites, are, therefore, metastable systems that take and keep their shape upon crystallisation and further calcination owing to a compromise between rigidity and flexibility of the various structural units. It is shown that, in the synthesis of porous silicates, one of the key effects that drive crystallisation is the framework ionicity in the as-made phase. This favours the formation of structural units which in the pure silica form are remarkably strained and barely stable. In this Perspective article, we employ ab initio quantum chemical results to analyse the influence of the polarisation of T–O bonds and, therefore, framework ionicity, in the synthesis of two types of zeolites: frameworks with cations more electropositive than Si and pure silica materials crystallised in the fluoride medium. It is shown that the enhancement of the structural flexibility driven by electronic effects in these cases favours the formation of structures with strained sub-units that in other synthesis conditions could not be reached.
 Evangelina Martínez-Morales | Evangelina Martínez obtained her Bachelor in Chemistry from the University of Guanajuato, México. She received her PhD in Physical Chemistry in 2007 at National Autonomous University of México, on a subject dealing with molecular dynamics simulations on zeolites. She is currently a post-doctoral fellow at Faculty of Sciences, Autonomous University of the Morelos State, México. She is currently involved in periodic ab initio calculations on hydrocarbon diffusion and structure directing effects on zeolites. |
 Claudio M. Zicovich-Wilson | Claudio Zicovich graduated as a Chemist at the University of Buenos Aires in Argentina and received his PhD in Physical Chemistry at the University of Valencia, Spain. Now he is full professor of Physics at the Faculty of Sciences of the Autonomous University of the Morelos State in Mexico. His area of research is the Quantum Chemistry of the Solid-State and he has made contributions to a wide variety of related topics: development and computational implementation of ab initio methods for crystalline solids, computational studies concerning heterogenous catalysis on oxide surfaces and porous materials, chemical reactivity of confined electronic systems, symmetry group theory, calculations of doped polymers and theoretical studies on processes of synthesis of porous silicates. Since 1995 he is part of the staff of developers of CRYSTAL, a public computational code which performs ab initio calculations of periodic systems. |
1 Introduction
Zeolites are porous alumino-silicates made up of TO4/2 units (T = Si, Al) that form 4-fold coordinated frameworks. These tetrahedral units are linked to each other by sharing their O-atoms so as to form structural rings, channels and cages in a 3-dimensional arrangement. Zeolitic frameworks appear, both in nature or by synthesis, in a large variety of microporous topologies1 with a wide compositional variability, and outstanding properties and attractiveness for a broad range of applications. The enormous synthetic efforts2 that have produced such a richness of zeolitic structures have initially relied on extensive trial-and-error research. However, as the empirical observations could be rationalised by means of key experiments3–5 it becomes less and less “blind” giving rise to a theoretical framework that allows to interpret and predict much synthetic processes.If only silicon atoms occupy framework T-sites, zeolites are silica polymorphs actually constituting metastable phases from the thermodynamic viewpoint. It has been shown6 that their formation enthalpy almost linearly depends on molar volume, being in general most favoured the densest materials. An important consequence is that, accordingly, the crystallisation of porous phases does occur against the thermodynamic trend that would drive the formation of α-quartz as the most dense and stable silica analogue.7 This may, in principle, suggest that the product selectivity takes place by kinetic control; however, one would keep in mind that the synthesised crystals are not actually pure silica polymorphs, but host–guest compounds. The presence of host molecules could influence the crystallisation process providing additional stability in order to yield highly porous materials as thermodynamic products.
An initial “viable nuclei” is also required to give the kinetic product, whose viability is chiefly related to the energetics of the host–guest compound. For instance, it has been shown that the stabilisation energies imparted by several organic Structure-Directing Agent (SDA) to different zeolite frameworks often crystallising under the same inorganic conditions are quite good indicators of the phase selectivity of the organic additives.8
In this respect, several so-called “structure-directing” effects that jointly determine the phase selectivity of a zeolite crystallisation have been identified. These effects can be adjusted, with more or less success, in seeking for new zeolitic structures with particular features. The use of organic compounds as SDA has been extremely fruitful in yielding medium and high-silica zeolites and there are some examples in which the organic cation was successfully modified to induce the crystallisation of new zeolites with predefined particularities.9,10
It is generally assumed that the structure-directing effect of the SDA molecules chiefly concerns their ability to efficiently fill the void regions inside the porous material. However, there are other less evident phenomena that could favour the crystallisation of particular topologies through the adoption of rather well defined synthesis conditions. For instance, it has been shown that, for fluoride mediated systems (see below), the degree of dilution of the synthesis gel may largely affect the output of the crystallisation: the less dense frameworks are apparently favoured as the concentration increases.11 This empirical observation, that has been recently termed the Villaescusa's rule, still lacks for a convincing rationale.12
On the other hand, the most frequently appearing secondary building unit in zeolitic topologies is the 4-member ring (4MR) one (see Fig. 1).1
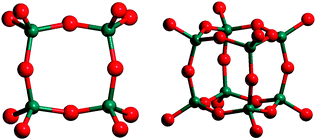 | ||
| Fig. 1 4MR (left) and D4R (right) secondary building units in zeolites. Red and green balls represent O and Si atoms, respectively. | ||
This significant occurrence takes place in zeolites with usually high Al content and the formation of these units might be associated with the ionicity induced by the presence of Al in T-sites. The presence of other heteroatoms like Ti, Ge or Ga in the framework would play a similar role to that of Al atoms, in particular in cages constituted by double-4MR (D4R) (see Fig. 1) or natriolite (NAT) units (see Fig. 2), which will be described in detail later in this paper.
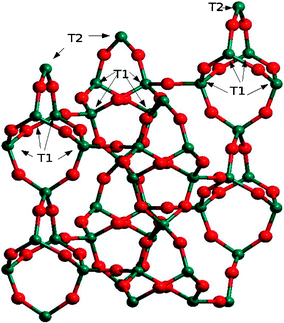 | ||
| Fig. 2 Structure of natriolite with T1 and T2 sites shown. Green and red balls correspond to Si and O atoms, respectively. | ||
In what follows we shall illustrate on how the electronic structure of the zeolitic framework plays a crucial role in directing synthesis towards structures that involve strained units. Depending on the degree of covalent character of the T–O bond, the resulting TO4/2 units exhibit a preference to keep the tetrahedron regularity, in association with the sp3 hybridisation of the T cation. When elements like Ge, Al, Ga, B, etc. occupy framework positions, the less electropositive the heteroatom is, the stronger such a preference and the more strained the framework will be. From the thermodynamical viewpoint, the purely silicic analogue, i.e. the less ionic, of a framework involving strained units like 4MR, D4R or NAT will be the least stable one.
In Section 3 we shall analyse in different cases how the electronic structure features related to the ionic character of the T–O bonds can be connected to the stability provided by the resulting structural flexibility.
Section 4 is devoted to show that a similar enhancement of ionicity of the T–O bonds takes place when performing the synthesis of silica zeolites along the F-route. In this case F− is used as mineraliser and the medium is typically close to neutral in contrast with the traditional hydrothermal route that requires basic medium. Furthermore, fluoride tends to get occluded into interstices of the silica framework and balance the charge of the organic cations. We shall show that, in this manner, topologies that are normally strained (and thermodynamically unfavourable) in the pure silica form become stable in the presence of fluoride and able to be crystallised. After precipitation, the F− and the organic SDAs are eliminated by calcination, yielding highly crystalline silica polymorphs remarkably free of defects.
Finally the way the F− anion is eliminated from the small cages inside which it resides in these strained as-synthesised silica zeolites is the subject of Section 5 where the mechanism of such a process is analysed in detail. The reaction that allows elimination provides additional evidences of the fact that the interaction between F− and the silica structure involves electronic effects that bring about a weakening of the Si–O covalent bonds in agreement with the previous results concerning ionicity.
2 General description of theoretical calculations
The following discussions are chiefly based on results obtained through quantum chemical calculations performed at high-level methods adopting the periodic hybrid-DFT (B3LYP13,14) approximation as implemented in the CRYSTAL code.15–17 This program makes use of the usual techniques of the molecular quantum chemistry by suitably adapting them to periodic systems. This is achieved thanks to the adoption of basis sets of Gaussian Type Atomic Orbitals which permits the exploitation of symmetry equivalences and localised character of the basis functions18 allowing for an efficient and feasible treatment of the infinite model.19 In particular, two different basis sets are mainly referred in this Perspective article: a valence double-ζ (VDZP) and a valence triple-ζ (VTZP) basis sets with polarisation functions, whose details have been reported in the cited works. The VDZP is the most commonly used one as it accurately describes the electronic behaviour of zeolitic frameworks with a relatively low computational cost. For a more detailed description of energetic features, the VTZP has been chosen at the expense of its high computational cost. In some specific cases, Grimme's-like dispersion correction20,21 for van der Waals interactions has been also considered together with the VTZP basis set. At this highest computational level, energy differences are also corrected for BSSE employing the counterpoise technique.22Finally, thermal effects and vibrational modes have been also computed considering the harmonic approximation. The structures considered have been optimised either to minima or transition states at the VDZP level by means of gradient techniques.15,23,24 In mechanistic studies the critical points have been topologically characterised through vibrational analysis.
In our experience electron densities, optimised geometries and relative stabilities of similar crystalline zeolitic systems are reasonably predicted at the VDZP basis set level using the hybrid-DFT approach. When comparing systems very similar from the compositional and structural point of view even the Hartree–Fock approach would give reasonable semiquantitative results. In what concerns the computation of vibrational modes, it has been shown25 that for silica and related materials the hybrid-DFT computational level provides good enough results even at not too high basis set level, as for instance the VDZP one. The accurate determination of electronic energies is the most tricky task for this kind of systems. In those cases a computational level that combines B3LYP hamiltonian, VTZP basis set, BSSE and Grimme's like corrections has been considered a reasonable choice to estimate energetics within chemical error for silicates.26
3 Presence of elements more electropositive than Si in the framework
Zeotypes are analogues to zeolites that exhibit elements other than Si, like P, Ga, B, Be, Zn, Ti, Ge, etc. in framework positions. The spatial distribution of these heteroatoms over the material depends on their preference for certain kinds of crystallographically distinct T-sites. This site-specific inclusion of heteroatoms drives in general the crystallisation toward microporous materials with specific topological features.12According to previous discussion, the formation of 4MR units in zeolitic frameworks could be associated in most cases with the ionicity induced by the presence of Al in T-sites. In what follows we shall show that the presence of other heteroatoms like Ti, Ge or Ga in the framework plays a similar role as Al atoms do, in particular in cages constituted by condensed 4MR: i.e.D4R or NAT units. This illustrates that not only extra-framework species like the well known organic templates play the role of SDAs but a significant driving force towards the crystallisation of particular topologies comes from chemical relaxation of the structures.
3.1 Intraframework germanium and zeolite flexibility
Let us start the discussion considering a case that does not actually involve 4MR units but exhibits features concerning the main topic of the present perspective article. Titanium-zeolites are employed as selective catalysts in oxidation reactions. In these materials Ti4+ are placed in framework T sites.27,28 The Ti content is rather low (<2%) and, owing to the Ti–O bond length larger than that of Si–O, 1.8 and 1.6 Å, respectively, the isolated Ti sites are quite strained as a result of its confrontation with the regular silica environment.In a quantum chemical study performed on periodic zeolitic models adopting the Hartree–Fock method with a valence double-ζ basis set,29 it has been shown that the isomorphic substitution of Si by Ge atoms in Ti-zeolites, particularly in the vicinity of the Ti atoms, contributes to stabilise the structure up to ∼17 kJ mol−1 per Ge/Si substitution. This effect has been attributed to the increase in ionicity of the framework provided by the presence of an intraframework cation more electropositive than Si. The larger ionicity is revealed by the Mulliken atomic charges on O atoms that in all considered cases increases in absolute value upon substitution of Si by Ge.
In the above-mentioned paper it has been argued that the increased framework ionicity should be associated with an enhanced structural flexibility, which promotes, at time, a more efficient relaxation around the Ti-sites. Remarkably, as revealed by the electron population analysis, the ionicity enhancement does not only take place on the O atoms in close vicinity with the T-sites occupied by Ge, but it propagates to further atoms decaying with distance. One of the main conclusions of the study29 is that the presence of Ge atoms occupying T-sites in the as-synthesised samples favours the inclusion of Ti atoms in the framework as a result of this ionicity–flexibility effect.
Similarly to the previous case, the presence of some heteroatoms in the framework appears to stabilise zeolites in which a structural strain is promoted by the occurrence of D4R units. While these units can only be formed in pure silica zeolites through the F-route of synthesis (see below), they are quite easily obtained when Ge atoms are incorporated in the framework. It is shown that the isomorphic substitution of Si by Ge increases the stability of zeolites with such units and favours their synthesis in the particular case of zeolite ITQ-7.30 Electronic structure calculations performed on cluster models indicate that the originally stressed O–T–O angles of the pure silica D4R units are relaxed owing to the preference of Ge to occupy D4R framework sites.30
The stability of the D4R units with respect to both species, in aqueous media and solid oxides, increases with the Ge content, from −134.3 and 131.8 kJ mol−1 for the unit without Ge atoms, to −177.0 and 80.8 kJ mol−1 for the substitution of Si by Ge atoms, respectively, in different corners of the cage. In other words, the 4MR imposes some degree of deformation of the TO4/2 units due to the small number of degrees of freedom allowed by the closure of the ring. Therefore, the less covalent the T–O bonds are, the more flexible is the TO4/2 unit and the less energetically demanding is its deformation, giving rise to a more stable structure. Hence the substitution of Si by Ge, a more electropositive cation, in D4R units enhances flexibility of the structure.
3.2 Gallium and natriolite units
Trivalent framework cations other than Al3+ give rise to an effect equivalent to that considered in the previous subsection. This is evidenced in a previous study of a peculiar behaviour in the crystallisation of a Ga-zeolite.31 The referred zeolite displays the NAT topology, as shown in Fig. 2 where two nonequivalent T-sites arise: T1 and T2. T1 comprises all corners of the “equatorial” 4MR of the NAT building unit and are connected to three other T1 and one T2 sites. The bridges between opposite corners of those rings are occupied by T2 sites, which are only connected to T1 sites. During the synthesis of this Ga-zeolite the transformation of an initially disordered phase into an ordered orthorhombic one was observed, while the disordered material approached a tetragonal phase even if it could not be synthesised as a fully tetragonal material. It was also observed that, for a Si/Ga ratio of 1.50, in the orthorhombic phase, T1 sites split into two sets of nonequivalent sites T1 and T′1 (Ga atoms only occupy the last one); while T2 sites are only occupied by Si atoms. Both effects are connected to the distribution of Si and Ga atoms in the NAT framework. It turns out that the presence of Ga in different framework positions has a structure-directing effect in the crystallisation of the zeolite.Taking these experimental results into account, the stability of one orthorhombic and three tetragonal models was calculated at the periodic hybrid-DFT level of calculations as implemented in the CRYSTAL03 code.32 From the point of view of the topological nature of the sites involved, the estimated energy cost of the exchange of a Si–Ga pair initially occupying sites T1 and T2, respectively, is about −7.5 kJ mol−1. Therefore, the structure with the lowest number of Si atoms located at T1 positions is the most stable one, i.e. the stabilisation provided by the substitution of Si by a more electropositive atom like Ga would be larger on T1 than T2.
On the other hand, the nature of the first neighbouring cations of each site determines the energetics of the exchange process, accordingly the energy cost for an exchange of a Ga atom on a site with a first neighbouring Ga has been calculated to be about 31.8 kJ mol−1. Compared with the cost of the Si–Ga exchange between T1 and T2, that of the exchange that introduces a trivalent cation in the close neighbourhood of another trivalent cation is about four times greater. Hence, it appears that the energetic preference of Si to occupy site T2 has a significant influence on the final cation distribution of the NAT topology with Si/Ga = 1.50.
The transformation of the disordered structure into the ordered one implies the breaking and formation of Si–O and Ga–O bonds, as well as the Ga and Si migration. Both phenomena take place in the mother liquor by a dissolution/recrystallisation process. The driving force for the recrystallisation appears to be the preference of Si to occupy T2 sites and, hence, the constraint of Ga to occupy only T1 sites without first neighboring Ga atoms. The disordered structure must be initially favoured because it is much more probable than the ordered one. However, the ordered phase is the most stable one since the number of Si atoms in T1 sites is minimised. Therefore, the initially formed disordered material evolves from the first stages of the synthesis toward the orthorhombic form.
Concerning the flexibility of the structure in the NAT topology, three O–T1–O bond angles are constrained within 4MRs, while two O–T2–O ones are in a similar situation.1 Therefore, the stabilisation provided by the substitution of Si by a more electropositive atom like Ga will be larger in T1 than in T2, due to the fact that the strain on the former will be larger than that of the latter. In this case the strain determined by the occurrence of condensed 4MR in the NAT unit drives the crystallisation of the system towards more flexible structures, as it occurred in the previous examples.
4 Synthesis through the F-route and D4R units in pure silica zeolites
Since the beginning of 1980's decade a novel technique for the synthesis of zeolites in which fluoride anions are employed as mineraliser in the crystallisation media has been more and more adopted as an alternative to the traditional hydrothermal route in a basic medium.33–35 This so-called F-route of synthesis has got growing success owing to: (a) the possibility to be performed in neutral or even slightly acid media as OH− ions are not actually required for the condensation of the T–O–T bridges, (b) the proved ability to yield materials with high Si content (or even purely silicic) and (c) the significant lack of defects in the resulting crystalline structures. An additional feature that makes this route of synthesis very appealing for industrial applications36 is that it allows to obtain pure silica porous materials with quite strained units like D4R that cannot be synthesised through the usual hydroxide route.This fact makes several authors to suggest that the presence of fluoride anions in the synthesis conditions plays a structure-directing role35 towards the formation of topologies exhibiting such strained units. However, up to a few years ago there was no convincing rationale able to support this hypothesis. In what follows, a few recent studies will be discussed showing that a genuine structure-directing effect toward the crystallisation of silica structures containing D4R cages is taking place during the synthesis in the F− medium. The enhancement of ionicity provoked by F− anions occluded in the framework cages is somehow similar to that considered in the previous section induced by framework Ge and Ga atoms.
4.1 Pure silica ITW: vibrational and electronic properties of the as-made and calcined samples
Pure silica ITQ-12 zeolite (ITW framework code1) is a highly crystalline material that can only be synthesised through the fluoride route with 1,3,4-trimethylimidazolium (TMI+) as SDA.36 The ITW framework consists of slit-shaped [44546484] obloid cages with six neighbouring D4R units. The obloid cages are in turn connected to each other through five-, six- and eight-ring windows. This gives rise to a bi-dimensional 8-members channel system which, together with the inert character of a defect free pure silica material, is well suitable for separation of gas mixtures constituted by small and very similarly sized molecules, like propane and propene.37,38Like in similar structures containing D4R cages, the F− anion is placed inside the ITW framework close to the centre of the cage in the as-synthesised sample.35 The TMI+, in turn, is located inside the slit-shaped obloid cage, which is efficiently filled owing to the planar conformation of the organic cation.36 The F− is eliminated from the material together with TMI+ during calcination yielding a practically defect free crystalline silica material.36 The ability of the anion to be eliminated from zeolites containing D4R cages at calcination temperatures and without structural connectivity breaking, despite it is tightly trapped inside the small cage, provoked some controversy in the literature.
Some authors39 claimed that the F− is not either actually located inside the cage or eliminated in the calcination process. This was based on the fact that computational simulations on Octadecasil zeolite showed that the energy barrier for the fluoride anion to pass through the 4MR window of the cage is about 3 eV, very high to allow diffusion at calcination temperatures.
On the other hand, accurate experimental data show that, for the same zeolite, the anion appears initially placed within the cage and it disappears in the calcined sample.40 The way such an elimination can occur will be the subject of discussion in Section 5. In what follows, we shall provide not only more evidences of the presence of F− inside the cages in the as-synthesised samples but also that such a presence is crucial in explaining the apparent structure-directing effect of fluoride towards the formation of D4R units.
In an analysis of the vibrational modes of as-made and calcined samples of SiO2-ITW, fingerprint peaks of the presence of F− in D4R cages have been characterised together with interesting evidences of non-trivial interactions between the framework and the occluded species in the as-synthesised material. The study has been performed by comparing IR and Raman spectra with accurate simulations of the same periodic structures carried out with the CRYSTAL06 code41 at the hybrid-DFT level of theory.42 The spectra and the calculated frequencies and intensities are shown in Fig. 3.
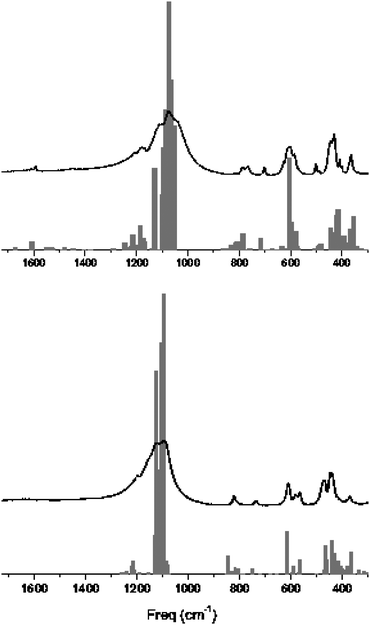 | ||
| Fig. 3 Experimental and simulated spectra for as-made (top) and calcined materials (bottom). Gray bars refer to B3LYP frequencies and intensities. Experimental spectra recorded at 298 K. Reproduced by permission from Zicovich et al.42 Copyright 2007 American Chemical Society. | ||
A very good agreement between experimental and simulated absorption peaks is exhibited, with maximum differences of about 20 cm−1 in some exceptional cases. This remarkable accuracy of the computational method adopted permits us to establish an unambiguous correspondence between theoretical modes and experimental peaks making possible the full assignment of the spectra and, furthermore, the analysis of important features of the nature of host–guest interactions and how they affect the flexibility and stability of the framework. For instance, a signal that neatly appears around 500 cm−1 in the as-made sample corresponds to an asymmetric breathing mode that in the absence of F− takes place at about 460 cm−1 in the calcined material. This shift is attributable to a decrease in the reduced mass owing to the additional contribution from the trapped anion. Similar results are obtained considering the Raman spectra.42
The possibility to make a detailed assignment of the vibrational spectra permits to analyse the richest and most complex part of them, which is the zone of lowest frequencies (<500 cm−1). In order to facilitate this analysis we shall make use of a mode decomposition into external/internal contributions of the Building Unit motions as discussed in ref. 42. In Fig. 4 the amount of external contributions to the motion of the SiO4/2 units is shown. This quantity gives an idea of the extent to which they behave rigidly during the motion.
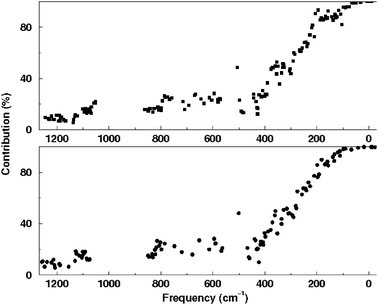 | ||
| Fig. 4 Total external contributions to the vibrational modes according to a partition in terms of SiO4/2 units. Bottom and top panels correspond to the clean (calcined) and as-made materials, respectively. Reproduced by permission from Zicovich et al.42 Copyright 2007 American Chemical Society. | ||
As it is shown in Fig. 4, the external contribution to the framework tetrahedral units decreases as the frequency increases, in a rather monotonous way, bearing resemblance to a sigmoid function. At the range of frequencies attributed to Si–O–Si bending and O–Si–O–Si torsions between different tetrahedral units, 200–500 cm−1, this contribution decreases quasi-linearly. At larger frequencies, comprising the O–Si–O bending and Si–O stretching regions, 550–900 and 1000–1280 cm−1, respectively, there is a slow decrease of contributions external to the tetrahedral units. As expected from previous studies43,44 there are several normal modes that practically behave as rigid unit modes with close to 100% of external motion of the tetrahedral units whose number should depend on the framework topology and, thereby, flexibility. The vibrational modes feature almost rigid tetrahedra motions at frequencies lower than 100 and 60 cm−1 for the clean and the as-made structures, respectively.
Of particular interest is that the fluoride contributions to the vibrational modes are maximal within the range of 100–160 cm−1, which is also a region where large differences between the SiO4/2 external contributions of the two systems appear. The SiO4/2 units of the as-made material display a less rigid behaviour in this zone with respect to the calcined one, exhibiting an apparent depression of more than 10% in the graphic shown in Fig. 4.
As the relative rigidity of the framework tetrahedral units is mainly due to the directionality of the SiO bond conferred by its partially covalent character, the increase in flexibility observed at 100–160 cm−1 in the as-made material is expected to be accompanied by an increase in ionicity of the SiO bond similar to that observed in the isomorphically substituted materials considered in Section 3. This suggests that the extraframework species in the as-made material exhibit an interaction with the zeolite inner surfaces that is much more complex than the short-range repulsion usually invoked to explain the structure directing action in terms of cavity filling. As a matter of fact, some extent of charge transfer does occur between these species and the framework in order to explain the vibrational behaviour. Owing to the relationship between IR intensity and dipole moment change, a relevant information about these electronic effects can be collected from the calculated vibrational spectra, which is validated by the good agreement with the corresponding experimental data shown in Fig. 3. It arises that the largest intensity values in the spectra, with frequencies in the range of Si–O stretching, i.e. 1000 and 1170 cm−1, are due to the significant change in dipole moment featured by the SiO bonds as the distance between the atoms increases on going towards heterolytic dissociation.
A much more surprising fact is that in the as-made material the difference in intensity between the SiO stretching band and the remaining modes lowers with respect to the clean material. This may be attributed to the participation of the charged TMI+ and F− in most modes in the low frequency region, which does increase their dipole moment change and, accordingly, the IR intensity. However, an analysis of the Born dynamic charges, here taken as the averaged diagonal elements of the ab initio computed Born tensor, suggests that a more complex phenomenon is possibly involved.
It is worth noting that, as a difference with the Mulliken partition of the electron density, the Born dynamic charges correspond to a macroscopic observable which provides the effective charges that should be attributed to each atom so as to reproduce the dipole moment changes brought about by small structural distortions. By definition the sum of all dynamic charges per cell must be zero.
Born charges computed for F− and TMI+ fragments as a whole are −1.20 and 1.43 |e|, respectively. While these values depend on a variety of electronic processes that take place during the atomic vibration (polarisation, electron transfer, etc.), it is remarkable that in both cases the dynamic charges exceed in magnitude the formal charges of the free ions. The only way this could occur is whenever the most probable motions of the ions are accompanied by a certain degree of charge transfer (or electron density polarisation) either toward or from the neighbouring atoms. In the present case, this means that the occluded ionic fragments dynamically exchange electrons with the framework atoms.
In the next section we shall show strong evidences that the presence of F− inside the D4R cage induces an apparent enhancement of ionicity of the Si–O bonds revealed in the present case in both Mulliken and Born atomic charges. This effect is at time reflected in a remarkable stabilisation of the silica zeolites containing strained units with condensed 4MRs like the D4R one.
4.2 Competitive crystallisation of pure silica TON and ITW
On the light of the previous analysis let us now consider a case in which two different zeolites can be formed in the crystallisation media along the F-route of synthesis: SiO2-TON and SiO2-ITW, shown in left- and right-hand sides of Fig. 5, respectively. These zeolites differ in two remarkable features: (1) the TON framework does not exhibit D4R cages and (2) it is denser than ITW. Both structural features allow to predict a larger stability of TON than ITW, in the absence of occluded species.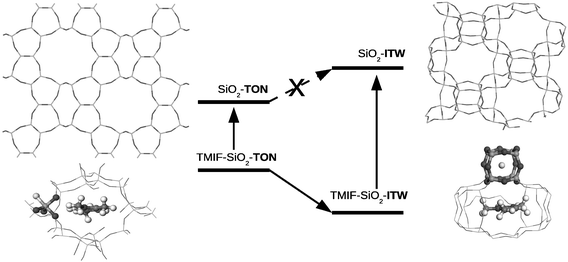 | ||
| Fig. 5 Schematic qualitative representation of the most relevant features of the competitive crystallisation of pure silica TON and ITW materials. Reproduced by permission from Zicovich et al.7 Copyright 2010 American Chemical Society. | ||
In the experiment performed with the same SDA (TMI+) as adopted for the SiO2-ITW synthesis,36TMIF-SiO2-TON initially crystallises without stirring, but after some time it transforms in situ into TMIF-SiO2-ITW.7 Both phases can be isolated pure at different crystallisation times in the same medium. Since the transformation occurs in situ, TON crystallises in this case through kinetic control and is transformed into ITW presumably due to the stabilising effect of both fluoride and TMI+ which overcome the predicted instability of its silica framework.7
The process can be schematically illustrated in Fig. 5. In a few words, while calcined SiO2-TON must overcome the framework instability to transform into SiO2-ITW, such a transformation easily takes place for the as-synthesised materials as the interaction with the occluded species is capable to revert their stability.
As it appears in Fig. 5, the way the extraframework ions are hosted in both silica structures differs owing to the structural particularities of each framework type. While in ITW, F− and TMI+ are located within the D4R and slit-shaped obloid cages, respectively (see Section 4.1), in TON, that lacks of such units, F− appears within a more open [6252] cage with a clear bonding to one of the Si atoms forming a [FSiO4/2]− pentacoordinated structure.7 Thus, the situation of F− within TON is much similar to the geometry predicted by Attfield et al.45 for [FSiO4/2]− units in other siliceous zeolites, and largely different from that found in ITW supported by XRD36 and calculations.42 Concerning TMI+, it is placed inside the 10-member channel in a partially disordered accommodation, being the most probable one a location parallel to the (101) plane of the primitive cell setting, as closest as possible to the [FSiO4/2]−group.7 The distribution of both ions brings about a less efficient cavity filling than in the ITW case.
In order to assess the relative stability of the different species intervening in the process, calculations performed with the CRYSTAL09 code15 at the hybrid-DFT level with a VTZP basis set have been considered, including a correction for van der Waals interactions20,21,26 and thermal contributions,7 as described in Section 2.
In what concerns the pure silica form of both zeolites (without occluded species) the calculated enthalpy of transformation from α-quartz agrees with the experimental correlation enthalpy vs. molar volume, provided in ref. 6, in that SiO2-TON is more stable than SiO2-ITW. However the computed enthalpy difference is larger than predicted by the molar volume correlation. This is to be attributed to the additional destabilising effect of the D4R cages in the ITW framework. The calculated value was ΔH298Ktrans = 5.49 kJ mol−1 per SiO2 formula unit, in contrast to the value of 1.54 kJ mol−1 obtained through the empirical correlation.
For the as-made materials, the following process has been considered,7
| TMIF-(SiO2)24-TON + (SiO2)12-ITW → 2(SiO2)12-TON + TMIF-(SiO2)12-ITW, |
The stabilisation provided by the occluded species to the as-synthesised zeolites can be separated into two different kinds of effects. On one hand, the larger stability of the most dense phase, SiO2-TON, is compensated by the cavity filling effect of the TMI+ that more efficiently match the slit-shaped obloid cage in ITW than in the 10-member channel of TON. This cavity filling effect has been shown to be mostly of long-range van der Waals nature.7
On the other hand, the energetic penalty imposed by the occurrence of D4R units in the ITW framework is overcame by the influence of F− inside the cage, that gives rise to a relaxation of the strained framework thanks to the increase of ionicity provoked by the anion, as discussed in Section 4.1. The different situation of the anion in both zeolites gives rise to different behaviours concerning the ionicity enhancement of the silica moiety. This can be observed in the distribution of differences in Mulliken atomic charges between the systems with and without occluded species depicted in Fig. 6.
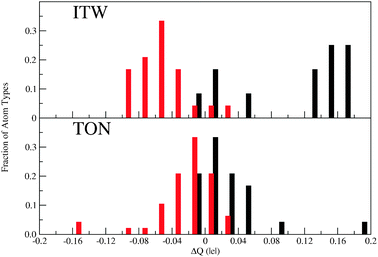 | ||
| Fig. 6 Distribution of Mulliken atomic charge differences between zeolites with and without occluded species, ΔQ, per framework and atom type. The fraction of atom types is computed within ranges of 0.02 |e| of ΔQ. Top and bottom panels correspond to ITW and TON, respectively. Black and red bars are associated to Si and O atom types, respectively. Reproduced by permission from Zicovich et al.7 Copyright 2010 American Chemical Society. | ||
In both TON and ITW the general trend is to increase the absolute values of the charges of Si and O in the presence of occluded TMI+ and F−, i.e. to increase the Si–O bond ionicity, in agreement with the discussion given in Section 4.1. However, the fraction of Si and O atoms that exhibits larger absolute charge differences, |ΔQ|, is much more significant in ITW than in TON, evidencing a remarkable global character of the effect in the former. Moreover, the fact that in the latter most atoms display quite small absolute charge differences and only a small fraction of 0.05 |e| of both atom types are affected by changes of more than 0.1 |e| indicates that in TON the Si–O bond polarisation is quite spatially localised, at variance with the ITW case.
While it may be surprising that the lack of a direct Si–F bonding has such an impact on the energetics and electronic distribution of the system, it turns out that the quite global Si–O bond polarisation mentioned above increases significantly the ionic character of the ITW framework. For a less covalent, less oriented Si–O bonding scheme, for which the rigid tetrahedral unit model no longer holds, the strain of the D4R units is partially released. On the contrary in TON the presence of F− just influences a few atoms of the framework causing drastic geometric and electronic changes closely around the Si atom to which fluoride is coordinated, while the remaining portion keeps the local features of the clean material.
This interpretation of the ionicity data, in connection with the framework flexibility, is also supported by the analysis of the irregularity of the SiO4/2 tetrahedra in each phase.7,42 The larger distortion of the tetrahedral units in ITW than in TON when they contain F− and TMI+ reflects the enhanced flexibility of the former.7 Additionally, the largest flexibility of those units that constitute the D4R cage, whose Si atoms also display the highest charge, suggests that the main cause for this additional flexibility of TMIF-SiO2-ITW is the F− occluded in the D4R unit.
5 Elimination of fluoride from the D4R cage in Octadecasil
It has been previously shown that the presence of F− inside the D4R cages of certain silica zeolites is crucial to allow the crystallisation of such materials. However, the experimental and theoretical evidences indicate that: (1) the anion is tightly trapped inside the cage in the as-synthesised sample, (2) diffusion across the 4MR windows is not possible in practice because of sterical effects,39,46 (3) at calcination temperature the anion is eliminated together with the SDA yielding a clean silica material with remarkably high crystallinity.It has been proposed40 that, as the fluoride elimination cannot proceed by simple diffusion (see Section 4.1), a more complex mechanism concerning hydrolysis and condensation of the SiOSi bridges of the cage might take place so as to explain the experimental results. Although a mechanism of this kind appears to be the only way to explain the experimental evidences, it still leaves some unsolved issues: (1) it is generally assumed that the formation of defects in a crystal is entropically favoured and the restoration to the perfect crystallinity should be a rather slow process, while experiments on fluoride elimination witness a fast recovery of the crystallinity upon fluoride removal, and (2) it is also assumed that pure silica zeolites are thermally resistant structures even at temperatures higher than that employed in the calcination, therefore, it seems unlikely at the first glance that the elimination of extraframework ions could involve the spontaneous breaking of the framework structure.
On the other hand, the studies discussed in the previous sections reveal that the fluoride anion is not merely residing inside the D4R cages but it experiments interactions with the neighbouring Si atoms which involve a non-negligible amount of electron transfer. Therefore a chemical mechanism of hydrolysis/condensation of the silica framework appears to be reasonable and, if verifiable, an additional support of the particular interaction between F− and the Si atoms in the D4R cage.
These considerations motivated a mechanistic study devoted to F− elimination from D4R cages. It has been carried out adopting the periodic approach as implemented in the already mentioned CRYSTAL code,15 at the B3LYP/VDZP//VTZP level of theory, including thermal effects and topological characterization of the critical points. The silica zeolite considered in this study was Octadecasil (AST code),1 being chosen because there are accurate and detailed experimental data on the fluoride elimination from this zeolite in the literature.40
The F-exclusion mechanism takes place in three steps, namely, (1) cage opening, (2) exiting of F− from the cage, and (3) cage closing. In the last step, the clean SiO2-AST structure is reached upon HF and H2O formation and further elimination from the zeolite. The notation adopted for the four stable species considered consists of two characters that indicate whether the cage is open (O) or closed (C) and two more to identify whether the F− is inside (i) or outside (o) the cage. Transition states are labeled denoting the features that change on going from reactants to products. They appear separated by colons, for instance, C:O denotes the transition state when going from a closed to an open cage. Accordingly, the label Oi refers to the species in which the cage is open while the F− is still inside it. For transition states, the label O(i:o) indicates the one exhibiting the cage opened with the fluoride evolving from inside to outside. The reaction profile together with schematic views of the different intermediates, including the D4R cage and the AST cavity, are shown in Fig. 7.
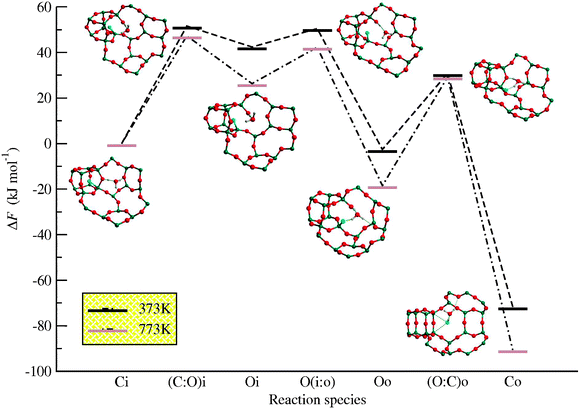 | ||
| Fig. 7 Helmholtz free energy reaction profile and schematic representation of the intermediates for the F-elimination from SiO2-AST. Reproduced by permission from Zicovich et al.47 Copyright 2010 American Chemical Society. | ||
The reaction is assumed to start from the F− inside the cage and a hydronium cation, H3O+, placed in the large cavity. The hydronium may appear as a result of the decomposition of the SDA at calcination temperatures. From a general inspection of the path it arises that the rate limiting step of the reaction is the first one, the cage opening, with a free energy barrier of 48 kJ mol−1 at calcination temperature (<773 K). In this step a SiO bond is broken by the concerted action of the hydronium that protonates the O-bridging and the F− that nucleophilically attacks the Si atom forming the typical [FSiO4/2]−group studied by Attfield et al.45 The quite low activation barrier of this step evidences the affinity of the anion to coordinate with the Si atoms in the D4R cage. This is coherent with the previously discussed studies (Section 4) that suggest the interaction of the F with the silica framework when occluded inside the cage involves a significant electronic reordering. On the other hand, the free energy results confirm that a hydrolysis of the SiO bond is absolutely feasible at calcination temperatures.
Another remarkable feature of the mechanism is that the F− is pulled out from the cage by forming a hydrogen bond with one of the water H atoms. The water molecule remains inside the Octadecasil cavity bounded to the silanol H atom. In this way a quite stable cyclic intermediate is formed (see Oo in Fig. 7). Unfortunately, the intermediate could not be experimentally characterised because it rapidly evolves toward products (SiO2-AST + HF + H2O) through a quite low free energy barrier of about 30 kJ mol−1 even at 373 K. The corresponding transition state is a 6-member structure through which, in a concerted way, the silica framework recovers the original connectivity. The occurrence of such an intermediate explains the fact that the final calcined sample keeps the original lack of defects of the as-made material.
In addition, the reaction free energy profile at a lower temperature (373 K) is also drawn in Fig. 7. It turns out that the same mechanism is also kinetically feasible at that temperature, requiring an appropriate weak acid medium. This is in agreement with indirect experimental evidences that in SiO2-β zeolite synthesised through the F-route, in which the anions are also occluded within D4R and other cages, the SDA and, presumably, the F− are eliminated at low temperature by treatment with aqueous acetic acid solutions.48
6 Concluding remarks
It has been shown by means of high-level computational analysis that the synthesis of silica zeolites through the F-route provides a way to enhance the ionicity in the framework in a similar way as the isomorphic substitution of Si by more electropositive cations (i.e.Ge or Ga) does. This enhancement of the ionic character of the SiO bonds favours the crystallisation of structures that in the pure silica form are barely stable from the thermodynamical viewpoint.In particular, we have analysed the effect of fluoride anions tightly occluded inside a small cage, like the D4R one, in the as-made materials. This unit, that consists of six condensed 4MR, is very strained when fully made of silica and is not stable enough to spontaneously form in nature or under alkaline hydrothermal laboratory conditions. The presence of F− appears to overcome this structural instability due to an interaction of electronic nature with the neighbouring Si atoms of the cage. This interaction involves some degree of nucleophilic attack to the Si atoms that in appropriate conditions (chiefly acid medium) can hydrolyse the SiO bonds of the cage and facilitate the elimination of F− from the cage. The calculated whole mechanism for such a reaction shows that the silica structure recovers the original connectivity thanks to the occurrence of a concerted step that drives the condensation toward the formation of the original SiO bonds.
From the physicochemical point of view this perspective review reveals that structure directing effects in the synthesis of zeolites appear to involve phenomena much more complex than the usually considered cavity filling of organic molecules used as SDA. While in the latter, short range repulsion and very weak attractive forces would play the main role, it arises that electronic effects may also be crucial in explaining the trends in the crystallisation of the different zeolitic phases during synthesis. For this reason, computational techniques that explicitly include the electronic structure of the intervening species at high-level of theory with realistic (periodic) models are often an important complement to experiments so as to acquire an accurate picture of the microscopic processes occurring in the synthesis of porous materials.
Acknowledgements
The authors kindly thank Miguel Camblor for fruitful suggestions and discussions concerning this topic and for providing most of the ideas developed in this paper. CZW also acknowledge the Crystal staff in Turin for the lasting and exciting collaboration of several years.References
- W. M. Meier and D. H. Olson, Atlas of Zeolite Structures, Butterworth, London, 1996 Search PubMed.
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663–702 CrossRef CAS.
- H. Gies, B. Marler and U. Werthmann, Molecular Sieves, Springer-Verlag, Berlin, 1998, vol. 1, pp. 35–64 Search PubMed.
- M. E. Davis and S. I. Zones, Synthesis of Porous Materials. Zeolites, Clays and Nanostructures, Marcel Dekker Inc., New York, 1996, pp. 1–34 Search PubMed.
- A. W. Burton, S. I. Zones and S. Elomari, Curr. Opin. Colloid Interface Sci., 2005, 10, 211 CrossRef CAS.
- P. M. Piccione, C. Laberty, S. Yang, M. A. Camblor, A. Navrotsky and M. E. Davis, J. Phys. Chem. B, 2000, 104, 10001 CrossRef CAS.
- C. M. Zicovich-Wilson, F. Gándara, A. Monge and M. A. Camblor, J. Am. Chem. Soc., 2010, 132, 3461–3471 Search PubMed.
- A. W. Burton, G. S. Lee and S. I. Zones, Microporous Mesoporous Mater., 2006, 90, 129 CrossRef CAS.
- S. I. Zones, M. M. Olmstead and D. Santilli, J. Am. Chem. Soc., 1992, 114, 4195 CrossRef CAS.
- L. A. Villaescusa, I. Díaz, P. A. Barrett, S. Nair, J. M. Lloris-Cormano, R. Martínez-Mañez, M. Tsapatsis, Z. Liu, O. Terasaki and M. A. Camblor, Chem. Mater., 2007, 19, 1601 Search PubMed.
- M. A. Camblor, L. A. Villaescusa and M. J. Díaz-Cabañas, Top. Catal., 1999, 9, 59 CrossRef CAS.
- M. A. Camblor and S. B. Hong, Porous Materials, Wiley, Chichester, 2011, pp. 265–325 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D. Arco and M. Llunell, CRYSTAL09 Users Manual, University of Turin, Turin, 2009 Search PubMed.
- R. Dovesi, R. Orlando, B. Civalleri, C. Roetti, V. R. Saunders and C. M. Zicovich-Wilson, Z. Kristallogr., 2005, 220, 571–573 CrossRef CAS.
- C. Pisani, R. Dovesi and C. Roetti, Hartree–Fock Ab initio Treatment of Crystalline Solids, Springer, Berlin, 1988, vol. 48 Search PubMed.
- C. M. Zicovich-Wilson and A. Erba, Theor. Chem. Acc., 2010, 126, 165 Search PubMed.
- Quantum-Mechanical ab initio Calculation of the Properties of Crystalline Materials, ed. C. Pisani, Springer, Berlin, Heidelberg, 1996, vol. 67 Search PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef.
- P. Jurecka, J. Cerny, P. Hobza and D. R. Salahub, J. Comput. Chem., 2007, 28, 555–569 CrossRef CAS.
- S. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553.
- B. Civalleri, P. D'Arco, R. Orlando, V. R. Saunders and R. Dovesi, Chem. Phys. Lett., 2001, 348, 131–138 CrossRef CAS.
- A. Rimola, C. M. Zicovich-Wilson, R. Dovesi and P. Ugliengo, J. Chem. Theory Comput., 2010, 6, 1341–1350 Search PubMed.
- C. M. Zicovich-Wilson, F. Pascale, C. Roetti, V. R. Saunders, R. Orlando and R. Dovesi, J. Comput. Chem., 2004, 25, 1873–1881 CrossRef CAS.
- P. Ugliengo, C. M. Zicovich-Wilson, S. Tosoni and B. Civalleri, J. Mater. Chem., 2009, 19, 2564–2572 RSC.
- G. Perego, G. Bellusi, C. Corno, M. Taramasso, F. Buonomo and A. Esposito, New developments in zeolite science and technology, Elsevier, Amsterdam, 1986, vol. 28, p. 129 Search PubMed.
- T. Blasco, M. A. Camblor, A. Corma, P. Esteve, J. M. Guil, A. Martínez, J. A. Perdigon-Melon and S. Valencia, J. Phys. Chem., 1998, 102, 75 Search PubMed.
- C. M. Zicovich-Wilson and A. Corma, J. Phys. Chem. B, 2000, 104, 4134–414 CrossRef CAS.
- T. Blasco, A. Corma, M. J. Díaz-Cabañas, F. Rey, J. A. Vidal-Moya and C. M. Zicovich-Wilson, J. Phys. Chem. B, 2002, 106, 2634–2642 CrossRef CAS.
- S. B. Hong, S.-H. Lee, C.-H. Shin, A. J. Woo, L. J. Alvarez, C. M. Zicovich-Wilson and M. A. Camblor, J. Am. Chem. Soc., 2004, 126, 13742–13751 CrossRef CAS.
- V. R. Saunders, R. Dovesi, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, N. M. Harrison, K. Doll, B. Civalleri, I. J. Bush, P. D. Arco and M. Llunell, CRYSTAL03 User's Manual, Università di Torino, 2003 Search PubMed.
- E. M. Flanigen and R. L. Patton, U.S. Patent 4 073 865, 1978 Search PubMed.
- P. Caullet, J. L. Paillaud, A. Simon-Masseron, M. Soulard and J. Patarin, C. R. Chim., 2005, 8, 245–266 CrossRef CAS.
- L. A. Villaescusa and M. A. Camblor, Recent Res. Dev. Chem., 2003, 1, 93–141 Search PubMed.
- X. Yang, M. A. Camblor, Y. Lee, H. Liu and D. H. Olson, J. Am. Chem. Soc., 2004, 126, 10403–10409 CrossRef CAS.
- D. H. Olson, X. Yang and M. A. Camblor, J. Phys. Chem. B, 2004, 108, 11044–11048 CrossRef CAS.
- P. A. Barrett, T. Boix, M. Puche, D. H. Olson, E. Jordan, H. Koller and M. A. Camblor, Chem. Commun., 2003, 2114–2115 RSC.
- A. R. George and C. R. A. Catlow, Zeolites, 1997, 18, 67–70 Search PubMed.
- L. A. Villaescusa, P. A. Barrett and M. A. Camblor, Chem. Mater., 1998, 10, 3966–3973 Search PubMed.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D. Arco and M. Llunell, CRYSTAL06 Users Manual, University of Turin, Turin, 2006 Search PubMed.
- C. M. Zicovich-Wilson, M. L. San-Román, M. A. Camblor, F. Pascal and S. Durand-Niconoff, J. Am. Chem. Soc., 2007, 129, 11512–11523 CrossRef CAS.
- K. D. Hammonds, V. Heine and M. T. Dove, J. Phys. Chem. B, 1998, 102, 1759–1767 CrossRef CAS.
- K. D. Hammonds, H. Deng, V. Heine and M. T. Dove, Phys. Rev. Lett., 1997, 78, 3701 CrossRef CAS.
- M. P. Attfield, C. R. A. Catlow and A. A. Sokol, Chem. Mater., 2001, 13, 4708–4713 CrossRef CAS.
- A. R. George and C. R. A. Catlow, Chem. Phys. Lett., 1995, 247, 408–417 Search PubMed.
- C. M. Zicovich-Wilson, M. L. San-Román and A. Ramírez-Solís, J. Phys. Chem. C, 2010, 114, 2989–2995 CrossRef CAS.
- C. W. Jones, K. Tsuji, T. Takewaki, L. W. Beck and M. E. Davis, Microporous Mesoporous Mater., 2001, 48, 57–64 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |