DOI:
10.1039/C0CY00096E
(Perspective)
Catal. Sci. Technol., 2011,
1, 354-365
Received
28th December 2010
, Accepted 31st January 2011
First published on 9th March 2011
Abstract
Six years have passed since renewed attention was paid to primary amines as potential organocatalysts in 2004. Numerous high quality studies in this field have provided chemists with valuable insights into the unique properties of chiral primary aminocatalysis. Among the diverse catalysts, natural cinchona alkaloid-derived catalysts have been widely regarded as a branch with high efficiency. This review examines the literature published between 2008 and late 2010, concerning cinchona alkaloid-derived primary amine catalysis.

Lin Jiang
| Lin Jiang was born in Kunming, China, in 1981. She received her BSc and MSc (Medicinal Chemistry, supervised by Professor Ying-chun Chen) from West China School of Pharmacy, Sichuan University in 2004 and 2007, respectively. In 2009, she joined the same research group as a PhD student. Her research interests are focused on the asymmetric reactions catalyzed by organic amines. |

Ying-Chun Chen
| Ying-Chun Chen was born in Chongqing, China, in 1972. He obtained his PhD from Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences (2001, Organic Chemistry). After working as a postdoctoral fellow in the same institute for one year, he joined Prof. Dan Yang’s group, in the Department of Chemistry, The University of Hong Kong, as a research assistant. In November 2003, he moved to West China School of Pharmacy, Sichuan University, and was appointed Full Professor in 2004. His research interests are in the areas of catalytic asymmetric organic synthesis, especially organocatalysis with chiral thiourea(urea) and amine compounds, and medicinal chemistry. |
1. Introduction
Catalytic asymmetric synthesis of optically pure compounds is one of the most prevailing focuses in the area of organic chemistry. Compared with traditional metal-based catalysts, chiral organic molecules as catalysts in a metal-free catalytic manner exhibit distinctive and competitive potency and intrigue synthetic chemists worldwide. The past decade has witnessed the rise and continuous prosperity of organocatalysis.1 In particular, the development of asymmetric aminocatalysis is at such a breathtaking pace that it has been known as a “gold rush in organic chemistry”.2 In light of the enormous amount of publications on asymmetric aminocatalysis, chiral secondary amines are undoubtedly the dominant catalysts for transformations of carbonyl compoundsviaenamine or iminium activation.3 However, recent studies4 revealed that encompassing the classic iminium activation mode of pyrrolidine-type catalysts, primary amines could overcome the inherent difficulties of secondary amines in generating congested covalent intermediates when more bulky carbonyl compounds are engaged, particularly in the case of α,β-unsaturated ketones or α-substituted α,β-unsaturated aldehydes.
Of all the classes of routinely used chiral primary aminocatalysts, those possessing cinchona alkaloid skeletons are perhaps the most remarkable performers,5 given that they are readily available, tunable and usually engage with concerted catalytic mechanism in asymmetric reactions.6 Since the pioneering reports by our group and Melchiorre in 2007, the catalytic capacity of cinchona alkaloid-based primary amines has been substantially upgraded by the efforts of many research groups. In order to restrict the topic to a more concentrated level and avoid overlap,5 this review will focus on the prominent asymmetric reactions which were catalyzed by cinchona alkaloid-derived primary amines between 2008 and late 2010.
2. Iminium catalysis
β-Functionalization of α,β-unsaturated ketonesvia chiral primary amine-mediated iminium catalysis has been considered as a more attractive strategy in comparison with that of secondary amines. The key intermediate in the catalytic system is the iminium ion generated by the condensation of primary amine and carbonyl compound under acidic conditions, which can serve as a reactive acceptor to participate in subsequent nucleophilic addition (Scheme 1).
The catalytic asymmetric Michael addition is generally regarded as one of the methods of choice in carbon–carbon bond-forming reactions,7 because a great diversity of nucleophiles could be expected to give versatile arrangements.
2.1.1
Carba-Michael addition reaction.
Sulfone functionality is usually employed as an electron-withdrawing-group to increase the electrophilicity or nucleophilicity of a parent reagent in organic synthesis. Jørgensen and co-workers8 reported the enantioselective Michael addition of β-keto-benzothiazol-2-yl sulfones 1 to cyclic α,β-unsaturated ketones 2. 9-Amino-9-deoxy-epi-quinine (I) was found to be the optimal catalyst to afford the key intermediates 3, which were useful precursors of derivatives containing divergent skeletal arrays. When aryl substitutions (R1) were attached,8a intermediates 3 could be transformed into alkynes 4 or alkenes 5. Besides, 1,5-diketo products 6 were also accessible viadesulfonylation. In contrast, when alkyl-substituted intermediates 3 were applied,8b an intramolecular aldol reaction together with subsequent Smiles rearrangement and elimination led to bicyclic products 7 (Scheme 2).
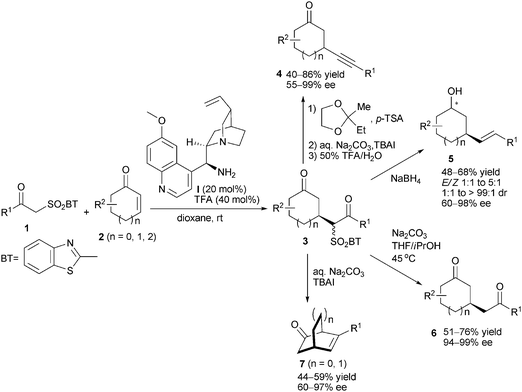 |
| | Scheme 2 Conjugated addition of β-ketosulfone and subsequent transformations. | |
Malononitrile
8 is an equivalent of 1,3-dicarbonyl compounds and the nitrile group is a versatile functional group that can be transformed into a variety of useful compounds. Silva et al. found that 9-amino-9-deoxy-epi-hydroquinine (II) in the presence of trifluoroacetic acid (TFA) was an effective catalyst for 1,4-conjugated addition of malononitrile 8 to 1,5-diarylpenta-2,4-dien-1-one 9. β-Regioselective products 10 were obtained exclusively, while no δ-adducts were observed (Scheme 3).9
α-Nitro
esters are good precursors of stabilized carbanions in organic synthesis. They have been used as masked α-amino acids in various organic transformations.10Luet al. demonstrated that a mixture of 9-amino-9-deoxy-epi-cinchonine (III) and (+)-camphorsulfonic acid (CSA) could efficiently catalyze the Michael addition of nitroacetates 11 to enones 12, affording the desired adducts 13 in excellent yields and up to 99% ee (Scheme 4).11Nitroalkanes are also good C-nucleophilic reagents. Yan and co-workers reported the conjugated addition of 1-bromonitroalkanes to α,β-unsaturated ketones catalyzed by amine (I).12
The indole framework is featured in numerous bio-active natural products and pharmaceutical agents. The inherent electron-rich characteristics make indoles valuable nucleophiles.13 Very recently, the Liu and Chen group14 reported that amine (I) could act as an efficient catalyst for the enantioselective nucleophilic substitution of cyclic Morita–Baylis–Hillman (MBH) products 14 with indoles 15. As summarized in Table 1, δ-substituted products 16 were obtained with exclusive regioselectivity and high enantioselectivity (up to 93% ee). The authors proposed that MBH products 14 firstly underwent a dehydration to form the iminium intermediates, then indoles 15 discriminately attacked the butadiene unit of the intermediates to generate optically active δ-products 16. Although the reactions afford substituted-type products, the transformation could be described as a conjugated-type addition when considering the reactive intermediate involved in the catalytic cycle.
Table 1 Asymmetric δ-regioselective substitution of cyclic Morita–Baylis–Hillman products 14 with indoles 15
| Entry |
R |
Indole
15 |
Yield (%) |
ee (%) |
A mixture of δ and γ products was obtained (δ/γ = 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35).
At 40 °C. :35).
At 40 °C.
|
| 1 |
H |
15a
|
16a-92 |
89 |
| 2 |
p-Br
|
15a
|
16b-88 |
93 |
| 3 |
m-Cl
|
15a
|
16c-80 |
92 |
| 4 |
H |
15b
|
16d-83a |
93 |
| 5b |
H |
15c
|
16e-70 |
47 |
| 6 |
H |
15d
|
16f-87 |
79 |
| 7 |
H |
15e
|
16g-90 |
86 |
2.1.2 Aza-Michael addition reaction.
Chiral amines are important building blocks in asymmetric synthesis. Aza-Michael addition to α,β-unsaturated carbonyl compounds represents one of the fundamental approaches towards this kind of valuable compounds.15 Although metal catalyzed asymmetric reactions have been reported on the part of enones, it was not until 2008 that Deng and co-workers established the first direct enantioselective aza-Michael addition to linear α,β-unsaturated ketones 12 (Scheme 5).16 Using Boc-protected N-benzyloxyamine 17 as nitrogen source, the conjugated addition could be catalyzed by the combination of amine (i.e.I) with TFA. They also deduced that while the chiral salt activated the enones 12viaiminium ion, the quinuclidine motif of (I), in either the free base (Scheme 5, mode a) or the protonated form (mode b), could bind to nitrogen nucleophiles such as 17viahydrogen-bonding interaction, thereby nucleophilic attack could happen due to the dual activation of the bifunctional cinchona alkaloid-derived primary amine. This protocol was applicable to a broad scope of α,β-unsaturated ketones, furnishing desired products 18 with excellent enantioselectivity.
 |
| | Scheme 5 Asymmetric aza-Michael addition of Boc-protected N-benzyloxyamine. | |
Pyrazole motif represents a key pharmacophore in many pharmaceutically active natural products or commercial drugs. Although 2-pyrazolin-5-ones 19 have been adopted as a carbon-centered nucleophile in organocatalytic Michael addition,17 Zhao and co-workers firstly applied compound 19 in chiral primary amine-mediated aza-Michael addition to aliphatic acyclic enones 12. Using benzoic acid as co-catalyst, β-(3-hydroxypyrazol-1-yl) ketones 20 with good yields and high enantioselectivity could be directly synthesized (Scheme 6).18
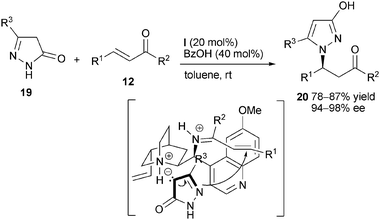 |
| | Scheme 6 Asymmetric aza-Michael addition of 2-pyrazolin-5-ones. | |
Wang et al. presented the procedure for enantioselective conjugated addition of a variety of N-heterocyclic compounds, such as 1H-benzotriazole 21 and 5-phenyltetrazole derivatives 23 (Scheme 7).19 The reactions could be smoothly promoted by employing amine (I) and benzoic acid. A variety of enones 12 could be used to provide Michael adducts 22 or 24 in high yield and with good to excellent enantioselectivity.
 |
| | Scheme 7 Asymmetric aza-Michael addition of various N-heterocyclic compounds. | |
2.1.3 Oxa-Michael addition.
Compounds possessing peroxide structural motifs usually have valuable biological activities due to the characteristic of peroxide to initiate radical reactions. Deng et al.20 reported the first example of enantioselective peroxidation of α,β-unsaturated ketones 12 catalyzed by primary amine (I). The ratio of peroxides 26 and epoxides 27 was found to be inversely correlated with reaction temperature. Enantiomerically enriched epoxides 27 were indeed obtained as the major products in moderate to high yields and excellent ee values at higher temperature (Scheme 8).
 |
| | Scheme 8 Enantioselective peroxidation of α,β-unsaturated ketones. | |
In an effort to expand the scope of the organocatalytic epoxidation, List and co-workers21 reported that catalyst (I) could efficiently catalyze the asymmetric hydroperoxidation of acyclic aliphatic α,β-unsaturated ketones 12 in the presence of trichloroacetic acid, furnishing enantioenriched cyclic peroxyhemiketals 29 in stable or unstable form. Compounds 29 could be easily transformed into epoxides 30 or β-hydroxy ketones 31 without ruining the ee values (Scheme 9).
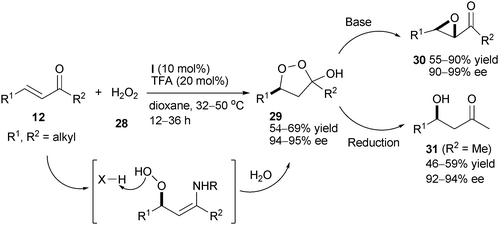 |
| | Scheme 9 Enantioselective hydroperoxidation of acyclic aliphatic enones. | |
2.1.4
Rearrangement reaction.
The development of synthetic methodology that can facilitate the construction of chiral quaternary carbon centers remains as one of the most challenging subjects in asymmetric catalysis.22 On the basis of their methodologies of stereoselectively constructing 1,3-diheteroatom units with 2-quaternary carbon centers via semipinacol rearrangement,23 Tu and co-workers24 reported the cinchona alkaloid-based primary amine-mediated semipinacol-type 1,2-sigmatropic migration with vinylogous α-hydroxy ketones 32, establishing the first organocatalytic enantioselective vinylogous α-ketol rearrangement reaction (Scheme 10). It was reasoned that in the presence of N-Boc-L-phenylglycine, 6′-hydroxy-9-amino-9-deoxy-epi-quinine (IV) could activate α-hydroxy ketones 32viaiminium ion resulting in an electron-deficient electrophilic center next to the tertiary hydroxyl moiety; subsequently, a semipinacol-type 1,2-carbon migration was induced, which led to the formation of an all-carbon quaternary stereogenic center. The spirocyclic diketones 33, which may serve as versatile building blocks in the synthesis of a number of natural products and pharmaceutical molecules, were furnished with good to excellent enantioselectivity.
 |
| | Scheme 10 Asymmetric vinylogous α-ketol rearrangementvia semipinacol-type 1,2-carbon migration. | |
Mechanistically, enamine activation of enolizable ketones starts with the formation of an iminium cation. The iminium intermediate can raise the acidity of the neighboring α-proton, thereby a deprotonation is triggered to form an enamine intermediate. In comparison with the original ketones, the nucleophilicity of the corresponding enamine intermediates are enhanced, thus α-functionalization of ketones can be realized through reactions with a variety of electrophiles.
3.1 Aldol reaction
The direct catalytic enantioselective aldol reaction is an important method in asymmetric carbon–carbon bond-forming reactions, since chiral β-hydroxy carbonyl compounds are in high demand in the synthesis of natural products and pharmaceutically active intermediates.25 The development of organocatalysis has reinvigorated this ancient reaction and abundant achievements have been accomplished in this research area.
In 2008, List and co-workers reinvestigated the enantioselective intramolecular aldolizations of 4-substituted 2,6-heptanediones 34 using cinchona alkaloid-derived primary amine catalysts.26 The combination of amine (I) with acetic acid turned out to be efficient for this reaction, giving highly enantiomerically enriched 5-substituted-3-methyl-2-cyclohexen-1-ones 35 (Table 2). The synthetic utility of this methodology was further exemplified by the first asymmetric synthesis of both enantiomers of celery ketone (35a, R = Me) under the catalysis of (I) or its pseudoenantiomeric analogue, 9-amino-9-deoxy-epi-quinidine (V). Both enantiomers are natural products and widely exist in plants. This work solves the long-standing problem of the enantioselective transformation of 4-substituted-2,6-heptanediones 34 to cyclohexenone derivatives 35.
Table 2 Asymmetric intramolecular aldolization of 4-substituted 2,6-heptanediones 34
| Entry |
R |
Yield (%) |
ee (%)a |
|
Reactions were catalyzed by amine (I).
|
| 1 |
Me |
35a-91 |
92 |
| 2 |
nPr
|
35b-94 |
90 |
| 3 |
n-C5H11 |
35c-96 |
93 |
| 4 |
Ph |
35d-93 |
91 |
| 5 |
m-ClC6H4 |
35e-92 |
92 |
| 6 |
2-Furyl
|
35f-95 |
94 |
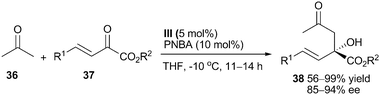
Very recently, another example of a simple ketone-involved aldol reaction was given by Chan and Kwong et al.27 They performed the reaction between acetone 36 and β,γ-unsaturated α-ketoesters 37 by the catalysis of amine (III) and p-nitrobenzoic acid (PNBA) (Scheme 11). This enantioselective protocol provided chiral aldol adducts 38 containing a quaternary carbon center together with tertiary alcohol, imparting excellent yield and enantioselectivity.
Enamine
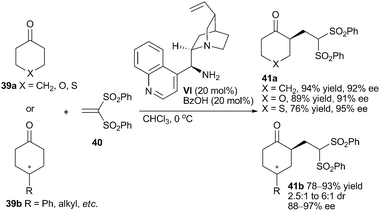
activation mediated Michael addition of ketones to various α,β-unsaturated systems is an attractive application of chiral primary aminocatalysts. Vinyl sulfone as a valuable electrophilic precursor has been introduced in more and more organocatalytic asymmetric reactions.28 In 2008, Lu and co-workers presented the Michael addition of cyclic ketones 39 to vinyl sulfone 40 by employing the mixture of 9-amino-9-deoxy-epi-cinchonidine (VI) and benzoic acid (Scheme 12).29 α-Alkylated carbonyl compounds and their derivatives 41 were afforded with generally satisfying yield and enantioselectivity.
4.
Dienamine catalysis
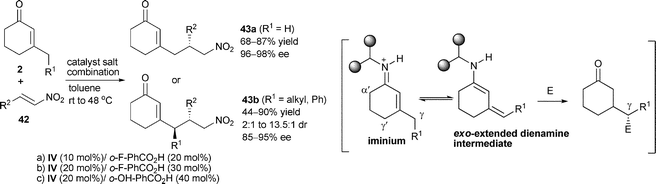
Dienamine catalysis developed by Jørgensen and co-workers provides a new activation mode in aminocatalysis,30 since it supplies an effective route to γ-functionalization of carbonyl compounds. In 2009, Melchiorre31et al. discovered the chiral primary amine-mediated dienamine catalysis, through which the direct intermolecular vinylogous Michael addition of unmodified β-substituted cyclohexenones 2 to nitroalkenes 42 could be smoothly promoted, providing exclusive γ-regioselective products 43 with high levels of diastereo- and enantioselectivity (Table 3). Both bifunctional catalyst (IV) and carboxylic acid co-catalyst were proved to greatly influence the reactivity and enantioselectivity. However, aliphatic nitroalkenes as well as the analogues of the nucleophilic component (i.e.3-methyl-2-cyclopenten-1-one) were inactive under optimal conditions, which suggested that the cyclic scaffold geometry strongly influenced the selective formation of the thermodynamic, exo-extended dienamine intermediate. Moreover, this protocol was equally applicable to β,β-disubstituted nitroalkene 42 or other Michael acceptors, such as compound 45 involving tandem vinylogous Michael addition/amination reactions, leading to compound 44 or 46 containing an all-carbon quaternary stereogenic center (Scheme 13). This elegant work confirms the unique advantages of chiral primary aminocatalysis and expands dienamine catalysis in the form of promoting vinylogous nucleophilicity within addition reaction manifolds.
 |
| | Scheme 13 Extended studies of the dienamine-catalyzed direct vinylogous addition. | |
| Entry |
Catalyst salt combination |
R1 |
R2 |
Yielda (%) |
dr |
Ee (%) |
|
If any, refers to the isolated single, major diastereoisomer.
Obtained after a single crystallization.
Refers to the isolated mixture of diastereoisomers.
|
| 1 |
a
|
H |
Ph |
43aa-77 |
— |
98 |
| 2 |
a
|
H |
4-MeO–Ph |
43ab-70 |
— |
97 |
| 3 |
a
|
H |
4-Br–Ph |
43ac-73 |
— |
97 |
| 4 |
a
|
H |
2-Thiophenyl
|
43ad-68 |
— |
98 |
| 5 |
b
|
Me |
Ph |
43ba-72 |
9:1 |
92 |
| 6 |
b
|
Me |
4-MeO–Ph |
43bb-78 |
10:1 |
94 |
| 7 |
c
|
Me |
4-NO2–Ph |
43bc-65 |
3:1 |
95 |
| 8 |
c
|
Allyl
|
Ph |
43bd-44 |
11.5:1 |
94b |
| 9 |
c
|
Ph |
Ph |
43be-86c |
2:1 |
90b |
Inspired by their recent discovery of the potential of chiral primary aminocatalysts in dienamine catalysis, Melchiorre and co-workers32 reported the direct asymmetric γ-alkylation of α-substituted linear α,β-unsaturated aldehydes 47 with bis(4-dimethylaminophenyl)methanol 48. The reaction was promoted by 6′-hydroxy-9-amino-9-deoxy-epi-quinidine (VII) with a chiral phosphoric acid (S)-49 as a co-catalyst (Table 4). The acidity of chiral phosphoric acids 49 strongly affected the reactivity, as it was crucial to induce in situ carbocation formation. Furthermore, as a hydrogen-bond donor, the hydroxyl group at the 6′-position of (VII) exhibited a positive influence on both the reactivity and stereoselectivity. A variety of γ-enolizable enals 47 possessing α-branched aliphatic substituent or phenyl group were applicable to this cooperative catalysis system which integrated dienamine catalysis and Brønsted acid catalysis, yielding SN1-alkylation products 50 with complete γ-regioselectivity and moderate to excellent ee values. This work serves as the first example of a catalytic asymmetric vinylogous substitution reaction of unmodified carbonyl compounds.
Table 4
Dienamine-catalyzed asymmetric γ-alkylation of α-branched γ-enolizable enals 47
| Entry |
R1 |
R2 |
Catalyst salt combination |
Yield (%) |
Ee (%) |
|
Reactions were carried out at 10 °C.
|
| 1 |
Bn
|
Me |
a
|
50a-82 |
90 |
| 2 |
Bn
|
Me |
b
|
50b-84 |
95 |
| 3 |
Allyl
|
Me |
c
|
50c-91 |
87 |
| 4 |
Allyl
|
Me |
b
|
50d-65 |
94 |
| 5 |
Et |
Et |
c
|
50e-58 |
82 |
| 6 |
Bn
|
Bn
|
c
|
50f-72 |
73 |
| 7a |
Ph |
Me |
a
|
50g-63 |
90 |
| 8a |
p-MeOC6H4 |
Me |
c
|
50h-72 |
86 |
5. Cascade reactions
Organocatalytic cascade reactions represent an attractive as well as highly efficient strategy for synthesis of optically active complex molecules. Numerous studies attributed to secondary aminocatalysis have highlighted the status of this reaction mode in the field of organocatalysis.33 Despite relatively few kindred studies grounded in primary aminocatalysis being done so far, it does capture considerable attention for a promising method of transforming bulky carbonyl compounds in a rational and convenient manner.
5.1.
Enamine-iminium catalysis
Spirocyclic oxindole skeletons exist in a large number of bio-active natural products. They are important building blocks in alkaloid synthesis and usually the pursuit of pharmaceuticals. However, the synthesis of spirocyclic oxindole motifs in an enantioselective way still remains as a challenging task.34 In 2009, Melchiorre’s group35 reported their work about asymmetric one-step construction of complex spirocyclic oxindoles by reacting olefinic oxindoles 51 with α,β-unsaturated ketones 12 under the catalysis of catalyst (II) (Scheme 14). The oxindole component 51 would first act as a Michael acceptor of the nucleophilic cross-conjugated dienamine intermediate. Then the resulting carbon nucleophile would selectively attack the β-site of enone moiety encompassing an iminium-catalyzed intramolecular conjugated addition to generate the desired spirocyclic oxindole derivative 52. The reactions proceeded well under the co-catalysis of o-fluorobenzoic acid and the compounds 52 featured with a spiro-oxindole moiety were furnished in moderate to high yield and generally high ee values. Especially, highly congested bicyclo[2.2.2]octanes 52a and 52b could be synthesized via the presented organocatalytic domino sequence, albeit in low yield.
 |
| | Scheme 14 Asymmetric construction of spirocyclic oxindolesvia double Michael addition. | |
Nearly at the same time, the same group36 applied the enamine-iminium catalytic strategy to the reactions of acyclic α,β-unsaturated ketones 12 with electron-deficient alkenes, such as nitroolefins 42, trans-α-cyanocinnamate 54 and maleimides 56, offering cyclohexane derivatives 53, 55 and bicyclic adducts 57 with excellent stereoselectivity (Scheme 15). This methodology complements the Diels–Alder reaction for the one-step synthesis of structurally complex cyclohexane scaffolds.
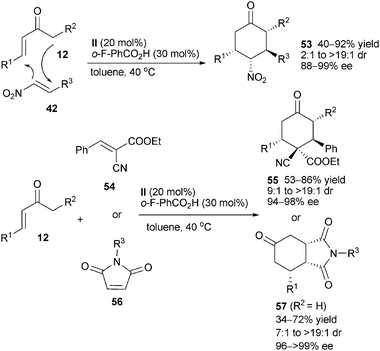 |
| | Scheme 15 Asymmetric organocascade reactions of enones with electron-deficient alkenes. | |
5.2. Iminium-enamine catalysis
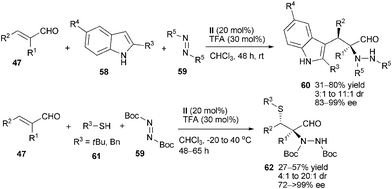
α,β-Disubstituted enals represent one of the challenging substrates in aminocatalysis. Melchiorre and co-workers firstly applied such substrates in chiral primary amine-catalyzed cascade three-component reactions.37 They demonstrated that catalyst (II) could selectively activate α,β-disubstituted enals 47 to undergo Friedel–Crafts/amination cascade sequence when using indole derivatives 58 as nucleophiles and azaodicarboxylates 59 as electrophiles. This method led to tryptophan derivatives 60 in moderate to good yield and high diastereo- and enantioselectivity. The cascade sulfa-Michael/amination sequence was also feasible on the premise that thiols 61 were used as nucleophilic components. The substrates 47 here showed good accommodation in terms of both electronic and steric effect, giving a variety of highly enantioenriched complex products 62 having a quaternary stereocenter contiguous to a C–S tertiary one (Scheme 16). This study supplies a convenient approach to optically active precursors of α-amino acids which have two adjacent stereogenic centers installing one quaternary carbon center.
 |
| | Scheme 16 Asymmetric cascade reactions of α,β-disubstituted enals. | |
Recently, Wang et al. described an organocatalytic straightforward approach to construct six-membered spirocyclic indoles (Scheme 17).38 α,β-Unsaturated ketones 12 and indole derivatives 63 underwent Michael-ketone aldol-dehydration domino sequence to afford desired products 64 in moderate to high yield and good to excellent enantioselectivity in the presence of catalyst (III) and TFA. The enones 12 showed broad substrate scope albeit ortho-substituents on the phenyl rings resulted in slight decrease in yield and enantiocontrol.
 |
| | Scheme 17 Asymmetric construction of spirocyclic oxindolesvia Michael-ketone aldol-dehydration domino reaction. | |
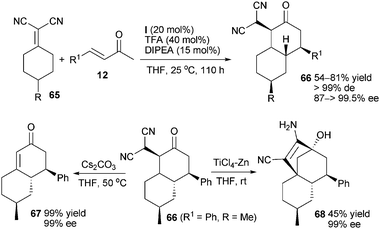
For the purpose of further exploring the vinylogous utilization of α,α-dicyanoalkenes,39 Chen et al. reported that catalyst (I) could efficiently promote the desymmetrisation of prochiral α,α-dicyanoalkenes 65via domino Michael–Michael addition to α,β-unsaturated ketones 12, giving chiral bicyclic compounds 66 bearing four stereocenters. The multifunctional bicyclic ketone 67 or tricyclic derivative 68 could be obtained by simple transformation of optically active product 66 without racemization (Scheme 18).40
 |
| | Scheme 18 Asymmetric desymmetrisation of prochiral α,α-dicyanoalkenes via domino double Michael addition. | |
5.3. [2 + 1] Annulation
5.3.1.
Epoxidation
.
Although epoxidation of olefins has proved to be a potent strategy to obtain synthetically useful chiral epoxides,41 it was not until 2008 when the first publication on highly eantioselective epoxidation of cyclic enones 2 was reported by the List research group.42 Employing hydrogen peroxide 28 as oxidant, the combination of primary amine (i.e.I) with proper acid (i.e.TFA) showed high catalytic efficiency, leading to enantiopure epoxides 69 in good yield and excellent ee values (Scheme 19).
A simple and general method for the direct catalytic asymmetric epoxidation of α-branched α,β-unsaturated aldehydes is urgently needed since α-substituted epoxyaldehydes are versatile synthons in organic synthesis. List and co-workers successfully extended their epoxidation strategy to α-branched α,β-unsaturated aldehydes 47 (Scheme 20).43 Their study revealed that the combination of a chiral cinchona alkaloid-based primary amine (i.e.I) with a chiral phosphoric acid (R)-49 (R = Ph or 2,4,6-iPr3C6H2) was crucial to gain high enantiocontrol. The authors proposed that, in addition to the primary aminocatalyst promoted iminium-enamine cascade sequence towards enals 47, the chiral phosphoric acid (R)-49 provided additional enantiodiscrimination in both steps, serving as a chiral counteranion (mode a) and as a Brønsted acid (mode b) respectively.
5.3.2. Aziridination.
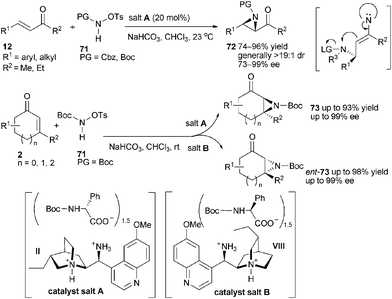
The development of catalytic asymmetric aziridination is of great value in view of the synthetic importance of aziridines.44 Recently, Melchiorre and co-workers reported the aziridination of linear enones4512 or cyclic46enones 2 (Scheme 21). The authors presumed that the choice of a suitable nitrogen-atom source was essential for developing the aziridination methodology. A desirable nitrogen-atom containing compound should first act as an N-nucleophile in iminium catalysis, affording a stereoselective conjugated addition step, and then become electrophilic to facilitate the enamine-catalyzed cyclization step. Further study revealed that the attachment of a good leaving group such as a tosyl moiety to N-protected reagent 71 could render the reaction biased towards a tandem sequence. Catalyst (II) or the pseudoenantiomeric primary amine, 9-amino-9-deoxy-epi-hydroquinidine (VIII) was found to be highly effective in the presence of chiral amino acid derivatives (catalyst salt A or catalyst salt B).47 These catalyst salts directed the reactions in a classic iminium-enamine catalytic manner towards N-protected acyclic aziridines 72 or cyclic ones 73 with almost complete diastereocontrol and generally very high enantioselectivity.
 |
| | Scheme 21 Asymmetric aziridination of enones. | |
5.3.3. Cyclopropanation.
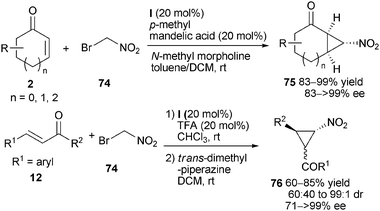
Cyclopropanes are synthetically important frameworks. Wang et al. reported that catalyst (I) could promote the enantioselective cyclopropanation of a variety of bromonitroalkanes 74 to cyclic α,β-unsaturated enones 2, yielding functionalized nitrocyclopropanes 75 with high levels of diastereo- and enantioselectivity (Scheme 22).48 It was proposed that the α,β-unsaturated enones 2 was first activated to suffer a nucleophilic conjugated addition via iminium catalysis, followed by an intramolecular alkylationviaenamine activation. The authors indicated that catalyst (I) could be used for kinetic resolution of cyclohexenone containing bulky substitutions at 4-positions. The enantioselective cyclopropane derivatives 76 derived from β-aryl-substituted enones 12 could be delivered while a two-step synthesis procedure was adopted.
 |
| | Scheme 22 Asymmetric nitrocyclopropanation of enones. | |
6. Cascade reactions based on hydrogen-bond activation
In contrast to the widely emphasized iminium or enamine activation, the less appreciated contribution of chiral primary amine-providing hydrogen bonding interaction with substrates, was employed by Zhong et al. in a stereoselective domino Michael–Henry reactions.49 Highly functionalized cyclohexanes 78 which contain four stereogenic carbon centers including two quaternary ones could be synthesized with high stereoselectivity. It was proposed that 1,3-dicarbonyl compounds 77 and nitroolefins 42 were synergistically activated by the tertiary amine moiety and primary amine moiety of catalyst (I) viahydrogen bonding interactions, thereby the conjugated addition was initiated. Subsequently, the carbanions derived from the Michael addition discriminatively attacked the carbonyl groups to afford the cyclic products 78. The same research group50 also applied this methodology to the enantioselective synthesis of multisubstituted cyclopentanes 79 (Scheme 23).
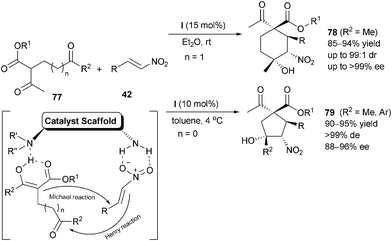 |
| | Scheme 23 Asymmetric synthesis of multisubstituted cyclic ketones through Michael–Henry reaction. | |
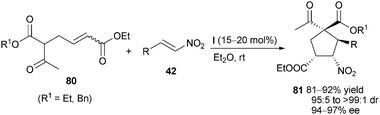
Catalyst (I) was proved to be able to efficiently catalyze the double Michael addition by Zhong and co-workers.51 The reaction was conducted between nitroolefins 42 and 5-acetyl-hex-2-enedioate 80 under mild reaction conditions, affording multisubstituted cyclopentanes 81 in high yield and excellent diastereo- and enantioselectivity (Scheme 24). This kind of activation protocol was also applied by them to the highly enantioselective Michael addition of 1,3-diaryl-1,3-propanedione to nitroolefins.52
7. Miscellaneous reactions
In the process of exploring new applications based on cinchona alkaloid-derived primary aminocatalysts, some other cascade reactions were reported. The key steps of these reactions are directed by either iminium activation or enamine activation.
Chromene derivatives have intrigued much interest in recent years because of their usefulness as bio-active substances and valuable synthons in the synthesis of many natural compounds. Xie et al. reported a convenient synthetic approach to optically active 2-amino-2-chromene derivatives.53 The salt of catalyst (I) here was employed as an iminium catalyst to promote Michael addition, followed by a cascade intramolecular cyclization, the chromene derivatives 83 or 84 were obtained with good to excellent enantioselectivity (Scheme 25). Later, they applied a similar strategy to the two-step synthesis of multifunctionalized tricyclic tetrazoles 86via intramolecular 1,3-dipolar cycloaddition as the key step (Scheme 26).54
 |
| | Scheme 25 Asymmetric synthesis of chromene derivatives through one-pot tandem reactions. | |
 |
| | Scheme 26 Asymmetric two-step synthesis of functionalized tricyclic tetrazoles. | |
A recent publication about asymmetric synthesis of functionalized 4-nitromethyl-chromans 88 was reported by Nugent and co-workers.55 The first step of this cascade reaction was induced by catalyst (I)-directed enamine activation. The chroman derivatives 88 could be generated in generally high yield and moderate to good ee values followed by an acid-catalyzed acetalization (Scheme 27).
 |
| | Scheme 27 Asymmetric synthesis of functionalized 4-nitromethyl-chromans. | |
8. Summary
Recent advances in asymmetric catalysis with natural cinchona alkaloid-based primary amines have been summarized in this review. The unique properties of these catalysts not only enable them to be broadly used in transformations of a variety of challenging carbonyl compounds which seem to be formidable substrates in chiral secondary amine catalysis, but also lead to superior reactivity and stereoselectivity over other types of primary aminocatalysts in some cases. The exploration in this area could be characterized into two styles, one of which is the discovery of new catalytic mechanisms, such as primary amine-directed dienamine activation, through which α,β-unsaturated carbonyl compounds could be selectively functionalized at the γ-position. The other one is characterized by the design of new reactions—especially new types of cascade reactions on the basis of iminium and/or enamine activation, which is viewed as a highly efficient strategy to access complex enantiopure compounds. Considering the inherent structural advantages as well as increased interest in catalytic potential at the current stage, we believe that the cinchona alkaloid-derived primary aminocatalysts will give rise to more promising findings in the near future.
Acknowledgements
We are grateful for the financial support from the NSFC (20772084).
Notes and references
- For general reviews on organocatalysis, see:
(a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS;
(b)
A. Berkessel and H. Gröger, Asymmetric Organocatalysis, Wiley-VCH, Weinheim, 2005 Search PubMed;
(c) B. List and J. W. Yang, Science, 2006, 313, 1584 CrossRef CAS;
(d) J. T. Mohr, M. R. Krout and B. M. Stoltz, Nature, 2008, 455, 323 CrossRef CAS;
(e) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS.
- For representative reviews on aminocatalysis, see:
(a) W. Notz, F. Tanaka and C. F. Barbas III, Acc. Chem. Res., 2004, 37, 580 CrossRef CAS;
(b) M. Marigo and K. A. Jørgensen, Chem. Commun., 2006, 2001 RSC;
(c) B. List, Chem. Commun., 2006, 819 RSC;
(d) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS;
(e) C. F. Barbas III, Angew. Chem., Int. Ed., 2008, 47, 42 CrossRef;
(f) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC.
-
(a) B. List, Tetrahedron, 2002, 58, 5573 CrossRef CAS; for recent reviews on iminium catalysis, see:
(b) G. Lelais and D. W. C. MacMillan, Aldrichimica Acta, 2006, 39, 79 CAS;
(c) A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 CrossRef; for recent reviews on enamine catalysis, see:
(d) B. List, Acc. Chem. Res., 2004, 37, 548 CrossRef CAS;
(e) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS.
- For general reviews on chiral primary aminocatalysts in organocatalysis, see:
(a) F. Peng and Z. Shao, J. Mol. Catal. A: Chem., 2008, 285, 1 CrossRef CAS;
(b) L.-W. Xu, J. Luo and Y. Lu, Chem. Commun., 2009, 1807 RSC.
- For representative reviews focused on cinchona alkaloid-derived primary aminocatalysts, see:
(a) Y.-C. Chen, Synlett, 2008, 13, 1919 CrossRef;
(b) G. Bartoli and P. Melchiorre, Synlett, 2008, 12, 1759.
- For comprehensive reviews related to organocatalysts possessing cinchona alkaloid structures, see:
(a) T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691 CrossRef CAS;
(b) S.-K. Tian, Y. Chen, J. Hang, L. Tang, P. Mcdaid and L. Deng, Acc. Chem. Res., 2004, 37, 621 CrossRef CAS ; see also Ref. 5. For the first synthesis of 9-amino-9-deoxy-epi-cinchona alkaloids, see: H. Brunner, J. Bügler and B. Nuber, Tetrahedron: Asymmetry, 1995, 6, 1699 Search PubMed.
- For recent reviews on organocatalytic conjugated additions, see:
(a) D. Almaşi, D. A. Alonso and C. Nájera, Tetrahedron: Asymmetry, 2007, 18, 299 CrossRef CAS;
(b) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701 CrossRef CAS.
-
(a) M. W. Paixão, N. Holub, C. Vila, M. Nielsen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2009, 48, 7338 CrossRef CAS;
(b) N. Holub, H. Jiang, M. W. Paixão, C. Tiberi and K. A. Jørgensen, Chem.–Eur. J., 2010, 16, 4337 CrossRef CAS.
- C. G. Oliva, A. M. S. Silva, D. I. S. P. Resende, F. A. A. Paz and J. A. S. Cavaleiro, Eur. J. Org. Chem., 2010, 3449 CrossRef CAS.
-
(a) G. Rosini and R. Ballini, Synthesis, 1988, 833 CrossRef;
(b) R. S. Fornicola, E. Oblinger and J. Montgomery, J. Org. Chem., 1998, 63, 3528 CrossRef CAS.
- C. Liu and Y. Lu, Org. Lett., 2010, 12, 2278 CrossRef CAS.
- L.-T. Dong, R.-J. Lu, Q.-S. Du, J.-M. Zhang, S.-P. Liu, Y.-N. Xuan and M. Yan, Tetrahedron, 2009, 65, 4124 CrossRef CAS.
- For selected organocatalytic examples, see:
(a) J. F. Austin and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 1172 CrossRef CAS;
(b) R. P. Herrera, V. Sgarzani, L. Bernardi and A. Ricci, Angew. Chem., Int. Ed., 2005, 44, 6576 CrossRef CAS;
(c) Q. Kang, Z.-A. Zhao and S.-L. You, J. Am. Chem. Soc., 2007, 129, 1484 CrossRef CAS;
(d) Y.-X. Jia, J. Zhong, S.-F. Zhu, C.-M. Zhang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2007, 46, 5565 CrossRef CAS.
- Z. Qiao, Z. Shafiq, L. Liu, Z.-B. Yu, Q.-Y. Zheng, D. Wang and Y.-J. Chen, Angew. Chem., Int. Ed., 2010, 49, 7294 CrossRef CAS.
- For recent reviews on enantioselective aza-Michael reactions, see:
(a) L.-W. Xu and C.-G. Xia, Eur. J. Org. Chem., 2005, 633 CrossRef CAS;
(b) M. Liu and M. P. Sibi, Tetrahedron, 2002, 58, 7991 CrossRef CAS.
- X. Lu and L. Deng, Angew. Chem., Int. Ed., 2008, 47, 7710 CrossRef CAS.
- S. Gogoi and C.-G. Zhao, Tetrahedron Lett., 2009, 50, 2252 CrossRef CAS.
- S. Gogoi, C.-G. Zhao and D. Ding, Org. Lett., 2009, 11, 2249 CrossRef CAS.
- J. Lv, H. Wu and Y. Wang, Eur. J. Org. Chem., 2010, 2073 CrossRef CAS.
- X. Lu, Y. Liu, B. Sun, B. Cindric and L. Deng, J. Am. Chem. Soc., 2008, 130, 8134 CrossRef CAS.
- C. M. Reisinger, X. Wang and B. List, Angew. Chem., Int. Ed., 2008, 47, 8112 CrossRef CAS.
-
(a) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363 CrossRef CAS;
(b) M. Bella and T. Gasperi, Synthesis, 2009, 1583 CrossRef CAS.
- For selected examples, see:
(a) E. Zhang, Y.-Q. Tu, C.-A. Fan, X. Zhao, Y.-J. Jiang and S.-Y. Zhang, Org. Lett., 2008, 10, 4943 CrossRef CAS;
(b) M. Wang, B. M. Wang, L. Shi, Y. Q. Tu, C.-A. Fan, S. H. Wang, X. D. Hu and S. Y. Zhang, Chem. Commun., 2005, 5580 RSC.
- E. Zhang, C.-A. Fan, Y.-Q. Tu, F.-M. Zhang and Y.-L. Song, J. Am. Chem. Soc., 2009, 131, 14626 CrossRef CAS.
- For recent reviews on catalytic asymmetric aldol reactions, see:
(a) S. Saito and H. Yamamoto, Acc. Chem. Res., 2004, 37, 570 CrossRef CAS;
(b) C. Palomo, M. Oiarbide and J. M. García, Chem. Soc. Rev., 2004, 33, 65 RSC.
- J. Zhou, V. Wakchaure, P. Kraft and B. List, Angew. Chem., Int. Ed., 2008, 47, 7656 CrossRef CAS.
- P. Li, J. Zhao, F. Li, A. S. C. Chan and F. Y. Kwong, Org. Lett., 2010, 12, 5616 CrossRef CAS.
- N. S. Simpkins, Tetrahedron, 1990, 46, 6951 CrossRef CAS.
- Q. Zhu, L. Cheng and Y. Lu, Chem. Commun., 2008, 6315 RSC.
- S. Bertelsen, M. Marigo, S. Brandes, P. Dinér and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 12973 CrossRef CAS.
- G. Bencivenni, P. Galzerano, A. Mazzanti, G. Bartoli and P. Melchiorre, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20642 CrossRef CAS.
- G. Bergonzini, S. Vera and P. Melchiorre, Angew. Chem., Int. Ed., 2010, 49, 9685 CrossRef CAS.
- For comprehensive reviews on organocatalytic cascade reactions, see:
(a) D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570 CrossRef CAS;
(b) X. Yu and W. Wang, Org. Biomol. Chem., 2008, 6, 2037 RSC.
- For selected organocatalytic examples, see:
(a) X.-H. Chen, Q. Wei, S.-W. Luo, H. Xiao and L.-Z. Gong, J. Am. Chem. Soc., 2009, 131, 13819 CrossRef CAS;
(b) Q. Wei and L.-Z. Gong, Org. Lett., 2010, 12, 1008 CrossRef CAS;
(c) K. Jiang, Z.-J. Jia, S. Chen, L. Wu and Y.-C. Chen, Chem.–Eur. J., 2010, 16, 2852 CrossRef CAS;
(d) K. Jiang, Z.-J. Jia, X. Yin, L. Wu and Y.-C. Chen, Org. Lett., 2010, 12, 2766 CrossRef CAS.
- G. Bencivenni, L.-Y. Wu, A. Mazzanti, B. Giannichi, F. Pesciaioli, M.-P. Song, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7200 CrossRef CAS.
- L.-Y. Wu, G. Bencivenni, M. Mancinelli, A. Mazzanti, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7196 CrossRef CAS.
- P. Galzerano, F. Pesciaioli, A. Mazzanti, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7892 CrossRef CAS.
- L.-L. Wang, L. Peng, J.-F. Bai, Q.-C. Huang, X.-Y. Xu and L.-X. Wang, Chem. Commun., 2010, 46, 8064 RSC.
- H.-L. Cui and Y. C. Chen, Chem. Commun., 2009, 4479 RSC.
- T.-R. Kang, J.-W. Xie, W. Du, X. Feng and Y.-C. Chen, Org. Biomol. Chem., 2008, 6, 2673 RSC.
- For a review on catalytic asymmetric epoxidation, see: Q.-H. Xia, H.-Q. Ge, C.-P. Ye, Z.-M. Liu and K.-X. Su, Chem. Rev., 2005, 105, 1603 Search PubMed.
- X. Wang, C. M. Reisinger and B. List, J. Am. Chem. Soc., 2008, 130, 6070 CrossRef CAS.
- O. Lifchits, C. M. Reisinger and B. List, J. Am. Chem. Soc., 2010, 132, 10227 CrossRef CAS.
- P. Müller and C. Fruit, Chem. Rev., 2003, 103, 2905 CrossRef CAS.
- F. Pesciaioli, F. De Vincentiis, P. Galzerano, G. Bencivenni, G. Bartoli, A. Mazzanti and P. Melchiorre, Angew. Chem., Int. Ed., 2008, 47, 8703 CrossRef CAS.
- F. De Vincentiis, G. Bencivenni, F. Pesciaioli, A. Mazzanti, G. Bartoli, P. Galzerano and P. Melchiorre, Chem.–Asian J., 2010, 5, 1652 CrossRef CAS.
- For the first example employing catalyst salt A, see: G. Bartoli, M. Bosco, A. Carlone, F. Pesciaioli, L. Sambri and P. Melchiorre, Org. Lett., 2007, 9, 1403 Search PubMed.
- J. Lv, J. Zhang, Z. Lin and Y. Wang, Chem.–Eur. J., 2009, 15, 972 CrossRef CAS.
- B. Tan, P. J. Chua, Y. Li and G. Zhong, Org. Lett., 2008, 10, 2437 CrossRef CAS.
- B. Tan, P. J. Chua, X. Zeng, M. Lu and G. Zhong, Org. Lett., 2008, 10, 3489 CrossRef CAS.
- B. Tan, Z. Shi, P. J. Chua and G. Zhong, Org. Lett., 2008, 10, 3425 CrossRef CAS.
- B. Tan, X. Zhang, P. J. Chua and G. Zhong, Chem. Commun., 2009, 779 RSC.
- J.-W. Xie, X. Huang, L.-P. Fan, D.-C. Xu, X.-S. Li, H. Su and Y.-H. Wen, Adv. Synth. Catal., 2009, 351, 3077 CrossRef CAS.
- X. Huang, P. Li, X.-S. Li, D.-C. Xu and J.-W. Xie, Org. Biomol. Chem., 2010, 8, 4527 RSC.
- D. B. Ramachary and R. Sakthidevi, Org. Biomol. Chem., 2010, 8, 4259 RSC.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.