Oriented irreversible immobilization of a glycosylated Candida antarctica B lipase on heterofunctional organoborane-aldehyde support
Melissa L. E.
Gutarra
ab,
Cesar
Mateo
a,
Denise M. G.
Freire
b,
Fernando A. G.
Torres
c,
Aline M.
Castro
d,
Jose M.
Guisan
*a and
Jose M.
Palomo
*a
aDepartamento de Biocatálisis, Instituto de Catálisis (CSIC), c/marie curie 2, cantoblanco campus UAM, 28049, Madrid, Spain. E-mail: josempalomo@icp.csic.es; jmguisan@icp.csic.es; Fax: +34-91-5854760; Tel: +34-91-585-4809
bInstituto de Química, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
cLaboratório de Biologia Molecular, Universidade de Brasília, Brazil
dGerência de Energias renováveis, Centro de Pesquisas e Desenvolvimento, PETROBRAS, Rio de Janeiro, Brazil
First published on 11th February 2011
Abstract
The immobilization of Candida antarctica (fraction B) lipase expressed in Pichia pastoris, a selective glycosylated protein at Asn 74, on a new heterofunctional support consisted of phenylboronic acid and aldehyde groups (Borald) has been performed. This method occurs via a two step mechanism: first orientation by organoborane interaction at neutral pH and a consecutive multi-point covalent attachment by aldehyde reaction at alkaline pH. The enzyme was specifically immobilized on this support in 70% yield at pH 7, oriented by the reaction of the hydroxyl groups on the sugar moiety with boronic acid on the support, whereas commercial CAL-B from Novozymes (non-glycosylated) was hardly immobilized at this pH (<10%). The consecutive incubation at pH 10 permitted the reaction of amine groups of the protein with aldehyde groups on the support. After a reductive amination, an irreversible immobilization methodology on organoborane-aldehyde support was possible with >99% final immobilization yield. The Borald-CAL-B preparation was a very stable biocatalyst in the presence of high amount of solvent or high temperature (e.g. more than 10 fold in the presence of 60% (v/v) acetonitrile). An improvement of the specific activity up to 5 fold for example in the hydrolysis of methyl phenyl acetate was obtained compared with a one-point covalent preparation. An ee of 89% towards R isomer was achieved with this new immobilized biocatalyst in the enantioselective hydrolysis of methyl mandelate.
Introduction
The immobilization of enzymes represents an exciting goal in the field of enzyme biotechnology.1–6Although apparently an older fashioned technique, immobilization has been revealed in the last times as a very powerful tool to improve almost all enzyme properties, if properly designed: e.g., stability, activity, specificity, selectivity or reduction of inhibition.7,8
Different strategies of immobilization such as ionic exchange, covalent attachment or hydrophobic adsorptions have been the most general established for preparation of supported biocatalysts.7,8 Other chemical selective methods based on the orientation of the protein by different groups, such as metal chelates or using cycloaddition reactions have been recently described.9,10
In particular, boronic acid molecular receptors for saccharides have attracted considerable interest due to their ability to bind saccharides in aqueous media.11–13
In addition, the immobilized boronic acids in gels can also be used for affinity chromatographic purification and detection of glycoproteins14,15 and oriented immobilization of glycoproteins,16–18 by their interaction with cis-1,2- or 1,3-saccharide diols to form five- or six-membered rings. Unfortunately, the interactions proved to be reversible and of very low affinity in aqueous media, rendering the immobilized biocatalyst of limited practical use.
Immobilization of enzymes by multi-point covalent attachment on highly activated support with aldehyde groups have represented an excellent irreversible methodology to create highly stabilized immobilized enzymes19 and even in some cases high enantioselective biocatalyst.20 The enzyme is immobilized at alkaline conditions through the lysine-rich regions.
Therefore, the design of a tailor-made heterofunctional support containing a combination of boronic acid groups and aldehydes is here proposed. These could permit the specific and oriented irreversible multi-point covalent attachment of glycosylated enzymes in aqueous media.21
This immobilization methodology involves two different steps: first, specific adsorption of the enzyme at pH 7 (where lysine moieties are not reactive with aldehyde groups) through a coordination of the hydroxyls groups of the sugar moiety with the boronic acid and second, the adsorbed enzymes are incubated under alkaline conditions to promote an intramolecular multi-point covalent attachment between aldehyde groups of the support and primary amino groups close to the adsorbed region of the enzyme (Scheme 1).
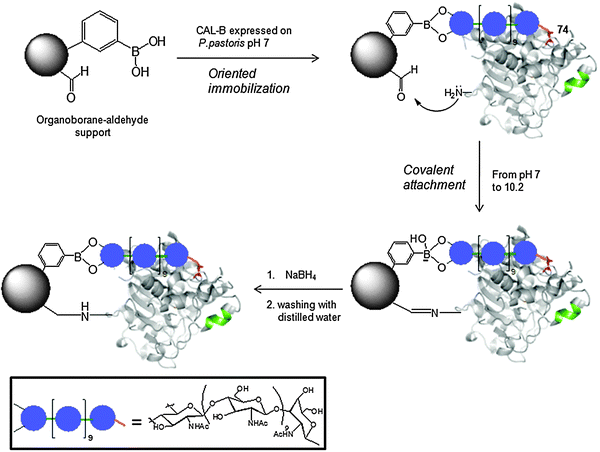 | ||
| Scheme 1 Oriented covalent immobilization of C. antarctica B lipase on heterofunctional organoborane support. | ||
Herein, the irreversible immobilization of Candida antarcticalipase B (CAL-B) expressed on Pichia pastoris, a site-specific glycosylated protein at Asn 74,22 using this novel protocol was studied. Firstly the thermal stability and the activity of the new preparation compared with a single point immobilized preparation (immobilized biocatalyst with the same properties of the soluble enzyme) were checked on the hydrolysis of four different esters: methyl butyrate (1), methyl phenyl acetate (2), methyl mandelate (3), 2-O-butyryl-2-phenylacetic acid (4) (Scheme 2). Finally, enantiospecifity of all preparations has been tested on the kinetic resolution of 3 and 4.
 | ||
| Scheme 2 Different esters hydrolyzed by CAL-B immobilized preparations. | ||
Results and discussion
Immobilization of CAL-B expressed in P. pastoris on organoborane-aldehyde support
Lipases from C. antarctica B expressed in P. pastoris and from Novozymes were immobilized on a heterofunctional aldehyde-organoborane support (Fig. 1).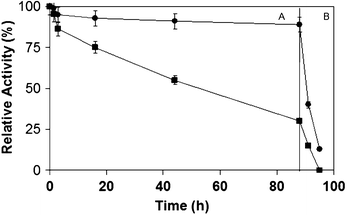 | ||
| Fig. 1 Immobilization profile of CAL-B expressed in P. pastoris versusCAL-B from Novozymes. CAL-B expressed in P. pastoris (squares), CAL-B from Novozymes (circles). Part A: immobilization at pH 7. Part B: incubation the previous preparation at pH 10. | ||
CAL-B from Novozymes was slightly immobilized on this heterofunctional support at pH 7, only around 10% in 80 h (Fig. 1). However, CAL-B expressed in P. pastoris, a selective glycosylated protein in Asn74 with a glucosamine oligosaccharide, was immobilized at 70% yield at pH 7. This demonstrated that this glycosylated CAL-B is specifically immobilized by the reaction of hydroxyl groups on sugar moiety with boronic acid groups on the support (Fig. 1).
Because this adsorption is reversible, the incubation of both enzymes at pH 10 permitted the multi-point covalent immobilization of the proteins after a reductive amination step.19CAL-B from Novozymes was immobilized almost 90% only when alkaline pH was used, therefore the enzyme in this immobilization is mainly oriented through the amino groups on protein (lysines) by reaction with aldehyde groups on the support, and not by the organoborane mechanism. CAL-B expressed in P. pastoris was immobilized with >99% yield. The glycosylation on this lipase allowed an oriented and irreversible immobilization on organoborane supports. A possible orientation of the enzyme during the immobilization is shown in Fig. 2, where it observes four lysines (for the multi-point irreversible covalent immobilization) around Asn74.
 | ||
| Fig. 2 Representation of the crystal structure of CAL-B. Lysines (blue), Asn74 (red), oligopeptide lid (green). The structure was obtained from the Protein Data Bank (PDB) using Pymolvs. 0.99. The pdb code for CAL-B is TCA. | ||
At pH 7 no immobilization of the enzyme was detected using supports activated with only aldehyde groups demonstrating that the immobilization was produced by boronate groups (data not shown).
In order to evaluate the scope of this methodology, the stability of the enzyme immobilized by this heterofunctional methodology was compared with a reference one-point immobilized preparation of CAL-B expressed in P. pastoris (glyoxyl-DTT-CAL-B), where the enzyme is one-point immobilized by the terminal amino group on the enzyme.23
The stability of the different enzyme preparations was studied at different conditions (Fig. 3). Particularly interesting was evaluating its application in complex green biotransformations were high temperature or the addition of high amount of co-solvent is necessary.
 | ||
| Fig. 3 Inactivation profile of different immobilized preparations of CAL-B expressed in P. pastoris. A. Incubation at 55 °C and pH 6. B. Incubation at 60% acetonitrile and pH 6. Organoborane-CAL-B preparation (squares), glyoxyl-DTT-CAL-B (circles). | ||
At 55 °C, CAL-B expressed in P. pastoris immobilized on Boronate-aldehyde support (Borald-CAL-B) maintained 50% of activity after 50 h, while the glyoxyl-DTT-CAL-B was completely inactivated. CAL-B supported on Borald was more than 7 fold stable compared with the one-point covalent attached enzyme by using the t1/2 of both enzyme preparations (Fig. 3A).
When this CAL-B preparation was incubated at 60% (v/v) acetonitrile, the effect was even clearer. The Borald-CAL-B preparation retained almost 60% activity after 80 h at these conditions whereas glyoxyl-DTT-CAL-B from P. pastoris was completely inactive after 25 h of incubation (Fig. 3B). Thus, CAL-B on this organoborane support was more than 10 fold more stable than the one-point covalent attached enzyme by the t1/2 analysis.
Activity and specificity of CAL-B expressed in P. pastoris immobilized on boronate-aldehyde support
The specific activity of the different preparations of CAL-B was evaluated in the hydrolysis of different esters (Scheme 2) at pH 7 and 25 °C (Table 1).The activity of the Borald-CAL-B P. pastoris preparation was between 3–5 fold higher compared with the glyoxyl-DTT-CAL-B preparation in the hydrolysis of 1, 2 and 3methyl esters at pH 7 (Table 1). However, the activity of Borald-CAL-B against 4 was 4 fold lower than that of the one-point covalent attached enzyme
The pH effect on the activity of CAL-B preparations was also evaluated in the hydrolysis of mandelic acid derivatives 3 and 4. When pH was changed from 7 to 5, both CAL-B biocatalysts underwent a decrease of 2 fold in the specific activity toward 3 whereas an enhancement of almost 3 fold in the activity of these biocatalysts toward 4 was observed.
Finally, the enantiospecifity of CAL-B from P. pastoris immobilized on Borald-support was evaluated on the kinetic resolution of 3 and 4 (Table 2). In both cases, the enzyme recognized mainly the R isomer.
The Borald-CAL-B preparation was the most enantiospecific immobilized biocatalyst toward both substrates and at two different pHs (Table 2). Better ee values were achieved compared with the glyoxyl-DTT-CAL-B preparation: from 79 to 86% at pH 5, or from 59 to 67% at pH 7 in the hydrolysis of 3 or from 39% to 58% in the hydrolysis of 4 at pH 7 (Table 2).
Conclusions
In conclusion, glycosylated CAL-B expressed in P. Pastoris has been specifically immobilized by oriented and irreversible method via a two step mechanism: organoborane formation and multi-point covalent attachment on heterofunctional organoborane-aldehyde support.This methodology permitted to obtain a very stable CAL-B biocatalyst, more than 7 fold in the presence of high amount of solvent or high temperature, an improvement on the specific activity up to 5 fold for example on the hydrolysis of 2, and a better selectivity in the enantioselective hydrolysis of 3, up to 89% ee towards R isomer.
Experimental
General
Candida antarctica lipase (fraction B) (CAL-B) was purchased from Novozymes. Agarose 10BCL was from Agarose Bead Technologies. Glyoxyl-agarose was prepared as previously described.19Methyl butyrate (1), phenylacetic acid methyl ester (2), methyl mandelate (3), 2-O-butyryl-2-phenylacetic acid (4), p-nitrophenylbutyrate (pNPB), epichlorhydrine, m-aminophenyl boronic acid, dithiothreitol (DTT) and sodium metaperiodate were purchased from Sigma Chem. Co. Other reagents were of analytical grade.Production of CAL-B in Pichia pastoris
The optimization of Candida antarcticalipase fraction B expression in Pichia pastoris was developed by the construction of a synthetic gene.24 The presence of a N-glycosylation site within the protein sequence at Asn 74 allowed glycosylation by P. pastoris.The C. antarctica B lipase expressed in P. pastoris was produced in agitated flasks. The inoculum was obtained by the propagation of one colony of P. pastoris in YPD (yeast extract 1%, peptone 2%, glucose 2%) and its incubation at 28 °C and 200 rpm for 15 h. 0.075 mg of cell (dry mass) from propagated inoculum was inoculated in 200 mL of YPD and incubated at 28 °C and 200 rpm for 48 h. At the end of fermentation, the culture medium was centrifuged at 5000 g and the supernatant stored at 4 °C.
Enzymatic activity assay
The activities of the soluble lipase, supernatant and enzyme suspension were analyzed spectrophotometrically measuring the increment in absorbance at 348 nm produced by the release of p-nitrophenol (pNP) (ε = 5.150 M−1 cm−1) in the hydrolysis of 0.4 mM pNPB in 25 mM sodium phosphate at pH 7 and 25 °C. To initialize the reaction, 0.05–0.2 mL of lipase solution or suspension was added to 2.5 mL of substrate solution in magnetic stirring. Enzymatic activity is given as one μmol of p-nitrophenol released per minute per mg of enzyme (IU) under the conditions described above.Purification of CAL-B
The enzyme was purified from commercial crude extract (CAL-B from Novozymes) or culture supernatant (CAL-B expressed in Pichia pastoris) by interfacial adsorption as previously described.25 Both enzymes were diluted in 10 mL of 5 mM phosphate buffer pH 7 (up to a final concentration of 0.005 mg of protein per mL) and the enzyme solution was added to one gram of octyl-agarose. The reaction was maintained for 2 h or until the immobilization was completed. After that, the suspension was filtered by vacuum and the solid was washed several times with distilled water. More than 95% of the enzyme was immobilized.For the preparation of the covalent immobilized catalysts, the lipase was desorbed from the support (one gram of octyl-CAL-B) adding 10 mL of a solution of 25 mM phosphate buffer pH 7 with 1% Triton X-100 (v/v) and incubated it for 2 h or until the desorption was completed. SDS-PAGE gel of both preparations reveals just one protein band (Fig. 4). A final solution of 5 μg purified lipase per mL was obtained.
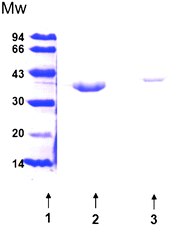 | ||
| Fig. 4 SDS-PAGE of purified CAL-B from Novozymes and expressed in P. pastoris. Lane 1: molecular weight marker. Lane 2: purified CAL-B from Novozymes (3 mg mL−1). Lane 3: purified CAL-B expressed in P. pastoris (0.05 mg mL−1). | ||
Preparation of the organoborane-aldehyde support
Ten grams of agarose 10 BCL was suspended in a mixture solution of 44 mL water, 16 mL acetone, 3.28 g NaOH, 0.2 g NaBH4 and 11 mL epichlorhydrine. The suspension was stirred mildly for 16 h and washed with an excess of water. After, these 10 g (epoxy-agarose support) were dissolved in 100 mL solution at pH 11 (100 mM sodium bicarbonate (80%) and dioxane (20%) (v/v)) with 5% (w/v) m-aminophenylboronic acid for 24 h at 25 °C.Periodate consumption of this support boronic acid-diol and another only with the same numbers of diols groups as reference were measured with similar results, dismissing a possible reaction between boronic acid and diols.
Periodate consumption was quantified using potassium iodide, as previously described.26 Finally the supports were oxidized adding a solution of 10 mL of water with 140 μmol of sodium periodate per gram of support during 90 min and washed abundantly with distilled water and store at 4 °C (Scheme 3).
 | ||
| Scheme 3 Synthesis of organoborane-aldehyde support. | ||
Immobilization of CAL-B on organoborane-aldehyde-support
One gram of organoborane-aldehyde support was added to 10 mL of purified CAL-B solution (0.005 mglipase per mL). Then, the suspension was stirred for 90 h at pH 7 and 25 °C. Periodically, samples of the supernatants and suspensions were withdrawn, and the enzyme activity was measured as described above. After that, the preparations were filtrated and re-suspended in 10 mL of 25 mM sodium hydrogen carbonate at pH 10.2 for 7 h. Finally, 10 mg of sodium borohydride were added (to reduce the imine bond to irreversible amine) for 30 min and then the immobilized preparation was filtrated and washed with distilled water (10 × 100 mL). The immobilization yield was >95%. Biocatalyst was prepared with a loading of 0.05 mg lipase per gram support.One-point covalent immobilization of CAL-B from P. pastoris
One gram of glyoxyl-agarose support was added to 10 mL of purified CAL-B solution (0.005 mglip per ml) with 50 mM DTT. Then the suspension was stirred for 2 h at pH 8 and 25 °C. Periodically, samples of the supernatants and suspensions were withdrawn, and the enzyme activity was measured as described above. Finally, the preparations were reduced by addition of 10 mg sodium borohydride for 30 min, filtrated and then washed with water. The immobilization yield was >95%. A one-point covalent attached preparation (glyoxyl-DTT-CAL-B) with a loading of 0.05 mglip per gsupport was prepared as reference catalyst for comparison (the immobilized preparation with similar properties than soluble enzyme).Enzymatic hydrolysis of different esters
The hydrolysis of 1 was performed by adding 0.3 g of immobilized enzyme to 10 mL of 10 mM of substrate in 10 mM sodium phosphate buffer at pH 7 and 25 °C. The hydrolysis of 2 was performed by adding 0.3 g of immobilized enzyme to 5 mL of 5 mM substrate in 10 mM sodium phosphate buffer at pH 7 and 25 °C. The hydrolysis of 3 was performed by adding 0.3 g of immobilized enzyme to 2 mL of 5 mM substrate in 10 mM buffer solution (sodium acetate at pH 5 or sodium phosphate at pH 7) at 25 °C. The hydrolysis of 4 was performed by adding 0.3 g of immobilized enzyme to 1 mL of 0.5 mM substrate in 10 mM buffer solution (sodium acetate at pH 5 or sodium phosphate at pH 7) at 25 °C.The degree of hydrolysis of 1–4 was analyzed by reverse–phase HPLC (Spectra Physic SP 100 coupled to an UV detector Spectra Physic SP 8450) on a Kromasil C18 column (15× 0.4 cm) supplied by Analisis Vinicos (Spain). At least, triplicates of each assay were made. The elution was performed with a mobile phase of acetonitrile (30%, v/v) and 10 mM ammonium phosphate (70%, v/v) at pH 2.95. The flow rate was 1 mL min−1. The elution was monitored by recording the absorbance at 225 nm.
Determination of enantiomeric excess
The enantiomeric excess (ee) of the produced acid (mandelic acid) was analyzed by Chiral Reverse Phase HPLC. The column was a Chiracel OD-R and the mobile phase was an isocratic solution of (5%, v/v) acetonitrile and (95%, v/v) 0.5 M NaClO4/HClO4 at pH 2.3 and the analyses were performed at a flow of 0.5 ml min−1 by recording the absorbance at 225 nm.Calculation of E-value
The enantiomeric ratio (E) was defined as the ratio between the percentage of hydrolyzed R and S isomers (from racemic mixture) at hydrolysis degrees between 10 and 20%, where the reaction kinetic is in first order. R- and S-isomers were used as standard enantiomerically pure products. Also the E value was calculated from the enantiomeric excess of the release acid (eep) and the conversion degree (c) using the equation E = ln[1 − c(1 + eep)]/ln[1 − c(1 − eep)] described by Chen et al.27Inactivation of CAL-B immobilized preparations against T and co-solvent
0.5 g of biocatalyst were dissolved in 5 mL of 25 mM sodium phosphate buffer (with 60% (v/v) acetonitrile) or incubated at 55 °C at pH 6. The remaining activity at different times was measured by the assay described above using pNPB as substrate.Acknowledgements
This work has been sponsored by the Spanish Ministry of Science and Innovation (project CTQ2009-07568), CSIC (Proyecto Intramural 200980I133) and CSIC (Proyecto Intramural 2008801058). Authors thank CNPq and Petrobras for financial support for Dr Gutarra.References
- W. Hartmeier, Trends Biotechnol., 1985, 3, 149–153 CrossRef CAS.
- A. M. Klibanov, Science, 1983, 219, 722–727 CrossRef CAS.
- E. Katchalski-Katzir and D. Kraemer, J. Mol. Catal. B: Enzym., 2000, 10, 157–176 CrossRef.
- L. Cao, L. van Langen and R. A. Sheldon, Curr. Opin. Biotechnol., 2003, 14, 387–394 CrossRef CAS.
- E. Katchalski-Katzir, Trends Biotechnol., 1993, 11, 471–478 CrossRef CAS.
- I. Chibata, T. Tosa and T. Sato, J. Mol. Catal. B: Enzym., 1986, 37, 1–24 Search PubMed.
- (a) C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS; (b) J. M. Palomo, Curr. Bioact. Compd., 2008, 4, 126–138 Search PubMed.
- (a) U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC; (b) J. M. Palomo, Curr. Org. Synth., 2009, 6, 1–14 Search PubMed.
- (a) F. Rusmini, Z. Zhong and J. Feijen, Biomacromolecules, 2007, 8, 1775–1789 CrossRef CAS; (b) P. Jonkheijm, D. Weinrich, H. Schroder, C. M. Niemeyer and H. Waldmann, Angew. Chem., Int. Ed., 2008, 47, 9618–9647 CrossRef CAS; (c) J. M. Palomo, Eur. J. Org. Chem., 2010, 6303–6314 CrossRef CAS; (d) D. Weinrich, P.-C. Lin, P. Jonkheijm, U. T. T. Nguyen, H. Schröder, C. M. Niemeyer, K. Alexandrov, R. Goody and H. Waldmann, Angew. Chem., Int. Ed., 2010, 49, 1252–1257 CrossRef CAS.
- (a) L. S. Wong, F. Khan and J. Micklefield, Chem. Rev., 2009, 109, 4025–4053 CrossRef CAS; (b) A. D. de Araujo, J. M. Palomo, J. Cramer, M. Kohn, H. Schroder, R. Wacker, C. Niemeyer, K. Alexandrov and H. Waldmann, Angew. Chem., Int. Ed., 2006, 45, 296–301 CrossRef CAS; (c) B. T. Houseman, J. H. Huh, S. J. Kron and M. Mrksich, Nat. Biotechnol., 2002, 20, 270–274 CrossRef CAS.
- T. D. James and S. Shinkai, Top. Curr. Chem., 218, 159 Search PubMed.
- W. Wang, X. Gao and B. Wang, Curr. Org. Chem., 2002, 6, 1285 CAS.
- S. Striegler, Curr. Org. Chem., 2003, 7, 81 CAS.
- B. M. Brena, F. Batista-Viera, L. Ryden and J. Porath, J. Chromatogr., A, 1992, 604, 109–115 CrossRef CAS.
- Y. C. Li, E. L. Larsson, H. Jungvid, I. Y. Galaev and B. Mattiasson, Chromatographia, 2001, 54, 213–217 CrossRef CAS.
- (a) N. Kanayama and H. Kitano, Langmuir, 2000, 16, 577–583 CrossRef CAS; (b) Bossi, S. A. Piletsky, E. V. Piletska, P. G. Righetti and A. P. F. Turner, Anal. Chem., 2001, 73, 5281–5286 CrossRef CAS.
- (a) A. Basso, P. Braiuca, S. Cantone, C. Ebert, P. Linda, P. Spizzo, P. Caimi, U. Hanefeld, G. Degrassi and L. Gardossi, Adv. Synth. Catal., 2007, 349, 877–886 CrossRef CAS; (b) R. Schoevaart, A. Siebum, F. Van Rantwijk, R. Sheldon and T. Kieboom, Starch/Staerke, 2005, 57, 161–165 Search PubMed.
- J. M. Abad, M. Velez, C. Santamaria, J. M. Guisan, P. R. Matheus, L. Vazquez, I. Gazaryan, L. Gorton, T. Gibson and V. M. Fernandez, J. Am. Chem. Soc., 2002, 124, 12845–12853 CrossRef CAS.
- (a) C. Mateo, J. M. Palomo, M. Fuentes, L. Betancor, V. Grazu, F. Lopez-Gallego, B. C. Pessela, A. Hidalgo, G. Fernández-Lorente, R. Fernández-Lafuente and J. M. Guisan, Enzyme Microb. Technol., 2006, 39, 274–280 CrossRef CAS; (b) C. Mateo, O. Abian, M. Bernedo, E. Cuenca, M. Fuentes, G. Fernández-Lorente, J. M. Palomo, V. Grazu, B. C. C. Pessela, C. Giacomini, G. Irazoqui, A. Villarino, K. Ovsejevi, F. Batista-Viera, R. Fernández-Lafuente and J. M. Guisan, Enzyme Microb. Technol., 2005, 36, 447–454 CrossRef.
- J. M. Palomo, G. Fernández-Lorente, C. Mateo, R. Fernández-Lafuente and J. M. Guisan, Tetrahedron: Asymmetry, 2002, 13, 2375–2381 CrossRef CAS.
- D. P. Gamblin, S. I. Van Kasteren, J. M. Chalker and B. G. Davis, FEBS J., 2008, 275, 1949–1959 CrossRef CAS.
- M. Jahic, J. Rotticci-Mulder, M. Martinelle, K. Hult and S.-O. Enfors, Bioprocess. Biosyst. Eng., 2001, 24, 385–393 Search PubMed.
- G. Volpato, M. Filice, M. A. Z. Ayub, J. M. Guisan and J. M. Palomo, J. Chromatogr., A, 2010, 1217, 473–478 CrossRef CAS.
- A. M. Castro, J. V. Bevilaqua, D. M. G. Freire, F. A. G. Torres, L. M. M. SantaAnna, M. L. E. Gutarra, C. A. Barbosa, R. V. Almeida, R. R. Menezes and A. G. Cunha, Processo de produção de lipases por meio de modificação genética de levedura. Pedido de Patente ao INPI: Privilégio e Inovação no. PI0905122-8 depósito em 17/12/2009, 2009.
- A. Bastida, P. Sabuquillo, P. Armisen, R. Fernandez-Lafuente, J. Huguet and J. M. Guisan, Biotechnol. Bioeng., 1998, 58, 486–493 CrossRef.
- T. P. Nevell, in Methods in Carbohydrate Chemistry, ed. B. Whistler, Academic Press, New York, 1963, vol. 3, pp. 210–225 Search PubMed.
- C. S. Chen, Y. Fujimoto, G. Girdaukas and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 7294–7299 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |