Nanokinetics for nanocatalysis
Dmitry Yu.
Murzin
*
Åbo Akademi University, Turku/Åbo, Finland. E-mail: dmurzin@abo.fi; Fax: +358 2215 4479; Tel: +358 2215 4985
First published on 1st March 2011
Abstract
The impact of nanoscience on heterogeneous catalysis is discussed with an emphasis on catalytic kinetics. Examples are presented demonstrating that the size of nanoparticles as well as the size of reacting molecules, which should be accounted for in the explanation of activity and selectivity.
 Dmitry Yu. Murzin | Prof. Dmitry Murzin studied chemical technology at the Mendeleev University of Chemical Technology in Moscow (1980–1986). He obtained his PhD in 1989 and his DSc (1999) at Karpov Institute of Physical Chemistry, Moscow. He worked at Universite Louis Pasteur, Strasbourg and Åbo Akademi University as a post-doc (1992–1994). Between 1995–2000 he was associated with BASF. Since 2000 he holds the Chair of Chemical Technology at Åbo Akademi University. He serves on several catalysis and chemical engineering editorial boards. He is co-author of a monograph, holds 3 patents and is a co-author of ca. 450 publications. |
Nanocatalysis
The catalysis community in the recent years is watching a widespread application of nanoscience (i.e. science of objects with smallest dimensions ranging from few nanometres to less than 100 nanometres) and nanotechnology (the purposeful engineering of matter at scales less than 100 nanometres to achieve size dependent properties and functions). Chemistry in general has for a long time dealt with joining atoms and group of atoms together with bonds on the sub-nanometer scale, and heterogeneous catalysis in particular has involved nanoparticles for more than a century, even industrially.1–5 Although the “nanoeffect” in many catalytic applications was considered for a long time and led to the introduction by Boudart of a concept of structure sensitive and structure insensitive reactions,6 widespread application of such characterization techniques as HRTEM brought renaissance to the cluster size effect.Particles of nanosized dimensions exhibit properties that are intermediate from the properties of atoms and the bulk material. Commonly utilized wet methods for preparation of such catalysts as supported metals in general result in a rather broad particle distribution, while utilization of colloidal chemistry approach7–9 might be able to provide a narrower cluster size distribution.
In recent years many dozens of studies appeared in the literature where comparisons of reaction rates (turnover frequencies) of materials with different size and shape in the nanometre range are given and sometimes even kinetic regularities (e.g. reaction orders, various selectivity aspects) are mentioned.10 Among studied reactions are hydrogenations, oxidation, decarboxylation, hydrogenolysis, Fischer–Tropsch synthesis to name a few.
In this current review a brief overview is given of some theoretical approaches that can be used to describe experimentally observed dependencies of kinetics and kinetic parameters on the cluster size and dimensions of reacting molecules.
1. Cluster size effect
Since catalysis is a kinetic phenomenon there is a natural interest in analyzing dependencies of the activity of catalysts as a function of metal or metal oxide particle size. The values of TOF are calculated per exposed surface site, thus structure sensitivity is due to changes of physico-chemical properties with a size decrease. Various explanations of structure sensitivity on a mechanistic level were proposed involving quantum size effects, metastability of nano-structures under reactions conditions, contribution of defect sites to catalytic activity, and involvement of metal-support interface.10 In addition, geometric (ratio of sites with different coordination environment), electronic (ionization potential, binding energies) or side (deactivation due to size dependent carbon deposition) effects could play a role.Few attempts can be found in the literature accounting for kinetic features of the cluster size effect based on a surface thermodynamic approach.11–14
In ref. 11–14 the chemical potential μ(r) or the surface energy excess of the metal atom in a metal particle with a radius of curvature r was supposed to be different from that in a metal particle of an infinite size (bulk-like) μ(∞) in the following way15,16
| μ(r) − μ(∞) = 2γΩ/r | (1) |
Thermodynamic functions of a small system differ from those of the corresponding macroscopic one17 as an additional term on an ensemble level should be added to a basic thermodynamic equation, namely the system (rather than molecule) chemical potential,18 which should be considered as an ensemble with adsorbed molecules.
In a conventional approach to treat chemical potential of adsorbed species or activated complexes,19 such potential is considered to be the same as for an unoccupied solid. When the size of a nanocluster is changing, no changes of the potential are expected independent of the presence or absence of an adsorbate, if the usual thermodynamic approach is applied. Within the framework of nanothermodynamics approach, it is expected that surface energy (or surface tension) will change upon adsorption in a complex fashion.20 A generalized case, when intrinsic (i.e. excess of surface energy with particle size increase) and induced (i.e. excess of surface energy due to external stress exerted by adsorbed molecules) contributions are present, is depicted on Fig. 1.
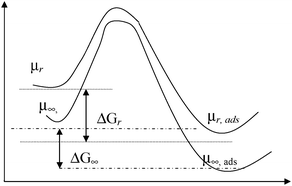 | ||
| Fig. 1 Potential energy diagram for adsorption when induced and intrinsic surface energy excess for nanoclusters. | ||
In ref. 21 it was supposed that not only the intrinsic chemical potential increment is inversely proportional to the cluster radius, but induced as well, moreover, the nature of adsorbed species, in particular the dimension of reactive molecules and the adsorption strength, was considered. This approach gives a possibility to extend the initial treatment given in ref. 11–14 to the case of multicomponent adsorption. Finally changes in the Gibbs energy (Fig. 1) can be described as below:
| ΔGads,∞ = ΔGads(r) + δ′solid/r − δ′solid,ads/r − δ′spe,ads/r = ΔGads(r) + Δδ′/r | (2) |
Taking into account relationships between the equilibrium constant and the Gibbs energy, as well as the relationship between thermodynamics and kinetics a following expression can be written for adsorption:21
| k(r) = gK(r)α = k∞eαη/r | (3) |
Quite often the two-step mechanism with two kinetically significant steps is used to describe catalytic kinetics:22,23
| (1) Z + A1 ↔ ZI + B1 (2) ZI + A2 ↔ Z + B2 A1 + A2 ↔ B1 + B2 | (4) |
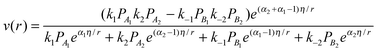 | (5) |
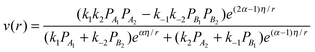 | (6) |
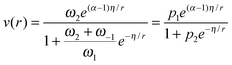 | (7) |
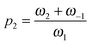 and p1 = ω2. Comparison of eqn (7) with experimental data was performed in ref. 13, 14, 21 and 24 demonstrating very good correspondence of theory and experiments. An example of fatty acids deoxygenation is illustrated on Fig. 2.
and p1 = ω2. Comparison of eqn (7) with experimental data was performed in ref. 13, 14, 21 and 24 demonstrating very good correspondence of theory and experiments. An example of fatty acids deoxygenation is illustrated on Fig. 2.
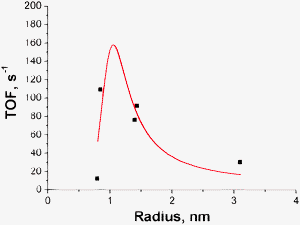 | ||
| Fig. 2 Dependence of TOF on the cluster radius in decarboxylation of stearic acid.24 | ||
The treatment above was extended21 for a Langmuir–Hinshelwood mechanism:
| (1) A + Z = ZA (quasi-equilibrium) (2) B + Z = ZB (quasi-equilibrium) (3) ZA + ZB ⇒ C + 2Z A + B → C | (8) |
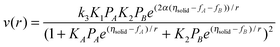 | (9) |
The framework of the thermodynamic concept discussed above can be applied for analysis of selectivity in for instance parallel reactions, when the first step is at quasi-equilibrium, while steps 2 and 3 are irreversible:
| (1) A + Z = ZA (2) ZA → B + Z (3) ZA → C + Z A → B; A → C | (10) |
The ratio between the rates is given by:
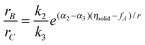 | (11) |
A more chemical explanation for the cluster size effect was discussed in ref. 25 where a cubo-octahedral representation was utilized and the fractions of edges to the total number of atoms on the surface, which includes besides edges also square and triangular faces, was calculated. The fraction of edges can be approximated by fedges ≈ 1/dcluster when d is given in nm. Such analysis allows introducing direct dependence of the cluster size into expressions of the rate constants. Since differences in the activation energy between edges and terraces are well recognized,26,27 in ref. 25 such difference in reactivity of edges and terraces was supposed to be responsible for cluster size effects. The theoretical concept advanced in ref. 25 leads to the same kinetic equations, as derived using a formal thermodynamic approach based on the changes of chemical potential.
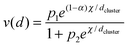 | (12) |
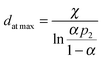 | (13) |
Although utilization of eqn (7) or (12) for analysis of TOF is very straightforward, a special care should be taken on which type of equation is valid for the reaction kinetics, as eqn (7) and (12) are based on two step sequence or Eley–Rideal mechanism, which is a special case of this sequence. Other types of mechanisms, for instance, Langmuir–Hinshelwood one might be valid for a particular reaction, thus in such case physico-chemical analysis of the values of kinetic parameters derived from eqn (7) could be misleading. In fact, Langmuir–Hinshelwood type of kinetic expressions could explain a maximum in the TOF dependence on the cluster size equally well as the two-step sequence. Thus, general evaluation of the cluster size effect should be combined with a proper kinetic analysis, when a form of a kinetic equation should be first established in a reliable way.25
Eqn (12) implies that there is a difference in catalytic activity between two types of sites. More real case should account for other types of active sites, present in heterogeneous catalysts. Another simplification of treatment above is the assumption that only one site is required for adsorption of one molecule.
2. Considerations of the molecules size
In recent years a lot of attention has focused on catalytic reactions where large organic molecules are involved. Historically, the development of kinetic approaches to catalysis was based on reactions involving small inorganic molecules, such as CO, oxygen, nitrogen, hydrogen, ethylene, etc. For such reactions it was logical to assume that just one surface site is required for a molecule to adsorb.It should be mentioned, however, that the size (cross section) of organic molecules often used in heterogeneous catalysis is ca. 0.5–1 nm, while the area occupied by small molecules like hydrogen is essentially smaller. The difference between the size of reacting molecules is rather seldom taken into account in kinetic modelling although several examples should be mentioned. Thus Frennet et al. studied the adsorption of saturated hydrocarbons and estimated the number of sites to be seven28 and eight or more,29 whereas Siffert et al.26 obtained a good description of the experimental data in 2-methylpentane skeletal transformation using a kinetic model with eight adsorption sites. Cabrera and Grau investigated methyl oleate hydrogenation and isomerization and found that cis- and trans-methyl oleate could cover up to eleven sites.30,31 In ref. 32 the number of Pd surface atoms needed for the adsorption of a single molecule of a natural lignan hydroxymatairesinol, extracted from wood knots, was elucidated to be equal to 20 (Fig. 3) by molecular modelling and this number was utilized in kinetic modelling.
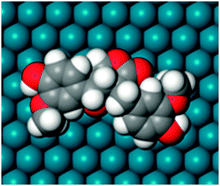 | ||
| Fig. 3 The molecule of hydroxymatairesinol adsorbed on palladium surface covering depending on the adsorption structure approximately 10–20 Pd surface atoms. | ||
As an area of ca. 1.6 nm2 is thus required for adsorption of hydroxymatairesinol and the size of a metal cluster is typically in the range of 1–4 nm, only few molecules could be adsorbed on the surface. Even for smaller reactants like carbohydrates or levoglucosan the kinetic diameter could be in the range of 0.5–0.7 nm, imposing a severe restriction on the number of adsorbed molecules per cluster.
In catalysis involving complex organic molecules with different functional groups, not only the number of sites, but also the mode of adsorption is important.
The influence of particle size on selectivity was discussed in relation to selective hydrogenation of α,β unsaturated aldehyde—cinnamaldehyde33 where larger metal particles give higher selectivity to cinnamyl alcohol. It was suggested, that on the largest metal particles the aromatic ring is not bound to the surface. The cinnamaldehyde molecule is tilted and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O will be closer to the surface with respect to the CC bond, leading eventually to higher selectivity.
O will be closer to the surface with respect to the CC bond, leading eventually to higher selectivity.
Another example is catalytic enantioselective hydrogenation of prochiral ketones.34 This reaction over heterogeneous catalysts produces racemic mixtures of enantiomers. When a chiral modifier e.g.cinchonidine in adsorbed on the catalyst surface the reaction becomes enantioselective favouring one product enantiomer over the other one. Adsorbed cinchonidine in parallel mode (active form) provides an enantioselective site and when the reactant is adsorbed in the vicinity of it, interactions between reactant and modifier lead to such orientation that hydrogenation to the main product is preferred. However, when the tilted form of modifier (spectator) is adsorbed in the vicinity of actor species the overall activity decreases. The parallel and tilted adsorption modes of cinchonidine require different amounts of primary Pt sites for adsorption, the former one occupying more active metal sites than the latter one. Kinetic modelling of 1-phenyl-1,2-propanedione hydrogenation took into account different number of sites, required for parallel and tilted adsorption, as well as site requirements for the reactants.35 Since an explicit equation for the rate cannot be derived equations for the rates of elementary steps were combined together with the mass balance for surface sites and solved numerically.
The same reaction of hydrogenation of 1-phenyl-1,2-propanedione was performed in the presence of cinchonidine with Pt/Al2O3 catalysts of different metal dispersion.34 The reaction rate was only slightly changed for catalysts with higher dispersion, at the same time enantioselectivity and to a certain extent regioselectivity was significantly varied. An optimum particle size in terms of enantioselectivity was ca. 4 nm. Initial increase of enantioselectivity with increase of metal size can be explained by the fact that for small particles of ca. 1 nm there is not enough space on the surface to accommodate the modifier (1.1 nm) in the parallel mode in addition to the reactant (0.7 nm). With increase of metal particle size enantio-and regioselectivity starts to decrease. One of the possible explanations for the lowering of selectivity at higher particle sizes is that the catalyst particle morphology changes becoming unfavourable for the enantiodifferentiation substrate–modifier interaction on the surface.
A semi-competitive model was developed in ref. 36 to account for the difference in size of reacting molecules assuming that the substrates do not absorb either with full competition, or with non-existing competition. For the liquid-phase reactions the catalyst surface is easily covered by these bulky organic molecules, while geometrical restrictions prevent organic molecules from complete covering of catalyst surface, implying existence of interstitial space between the larger organic species adsorbed on the surface sites. On such places smaller (by comparison) atoms (hydrogen or oxygen) are able to adsorb.
Kinetic treatment of such reactions should invoke the concepts of multi-centred species and incorporate several sites for an organic molecule and one (or two) site(s) for hydrogen (oxygen) activation with partial competition between the reactants for such sites.37 A complication with such models is in the numerical treatment, as solving the system of differential equations together with an algebraic one might be challenging.
3. Number of molecules per cluster
Kinetic equations, discussed in section 1, give a possibility to explain size dependent kinetics on a quantitative level without considering, however, the size of reacting molecules, which was addressed in section 2.An alternative approach for describing catalytic kinetics without introducing an explicit dependence on the cluster size is to consider the exact number of molecules, which can adsorb on a nanoparticle.38 Such approach demonstrated that the local coverage of adsorbed species on clusters of a particular size follows the Langmuir isotherm if the adsorption constant is independent of the amount of occupied molecules in a cluster. For a case when there is a distribution of clusters with different number of ensembles in each, the global coverage is a summation of the occupation of a particular cluster corrected by the probability of occurrence of this cluster. When adsorption constants are independent on the cluster size Langmuir adsorption isotherm can be obtained. This concept was later on39 extended to a two step catalytic sequence.
In case of the mechanism:
(1) Z + A ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) ZI ZIZI → Z + B A ↔ B | (14) |
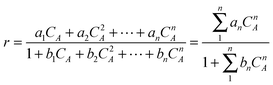 | (15) |
This equation represents a case of non-ideal surfaces when all kinetic and adsorption constants depend on the spatial arrangements of reacting molecules mainly due to lateral interactions between them.
In the simplified case of the equality of all adsorption and kinetics constants eqn (15) can be transformed to:
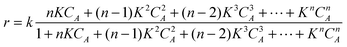 | (16) |
Final remarks
A classical kinetic approach to heterogeneous catalytic reactions often neglects the dimensions of clusters as well as the reacting molecules. For the majority of organic catalytic reactions over nanometre-sized transition metal clusters this is a very crude approximation. Experimental data discussed in the present review demonstrate that kinetic modelling on catalytic reactions over nm-size metal particles, i.e. nanokinetics, should include both the size of reactants and products, as well as the size of catalyst clusters.Future kinetic analysis could consider such important topics as cluster size dependent deactivation, influence of lateral interactions, extension of the current treatment to more complex mechanisms, inclusion of the particle size distribution as well as the impact of not only the size but also the shape of the nanoclusters.
References
- A. T. Bell, Science, 2003, 299, 1688 CrossRef CAS.
- G. M. Whitesides, Small, 2005, 1, 172 CrossRef CAS.
- N. Pernicone, CATTECH, 2003, 7, 196 CrossRef CAS.
- R. Schlögl and S. B. Abd Hamid, Angew. Chem., Int. Ed., 2004, 43, 1628 CrossRef.
- G. C. Bond, Acc. Chem. Res., 1993, 26, 490 CrossRef CAS.
- M. Boudart, Adv. Catal., 20, 153 Search PubMed.
- G. A. Somorjai and J. Y. Park, Angew. Chem., Int. Ed., 2008, 47, 9161 CrossRef.
- R. Narayanan and M. A. El-Sayed, Top. Catal., 2008, 47, 15 CrossRef CAS.
- G. A. Somorjai and J. Y. Park, Top. Catal., 2008, 49, 126 CrossRef CAS.
- F. Klasovsky and P. Claus, in Metal Nanoclusters in Catalysis and Materials Science: The Issue of Size Control, ed. B. Corain, G. Schmid and N. Toshima, Elsevier, Amsterdam, 2008, pp. 167–181 Search PubMed.
- V. N. Parmon, Dokl. Phys. Chem., 2007, 413, 42 CrossRef CAS.
- V. N. Parmon, Thermodynamics of Non-Equilibrium Processes with a Particular Application to Catalysis, Elsevier, Amsterdam, 2010 Search PubMed.
- D. Yu. Murzin, Chem. Eng. Sci., 2009, 64, 64.
- D. Yu. Murzin, J. Mol. Catal. A: Chem., 2010, 315, 226 CrossRef CAS.
- S. Parker and C. T. Campbell, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75 Search PubMed.
- C. T. Campbell, S. C. Parker and D. E. Starr, Science, 2002, 298, 811 CrossRef CAS.
- T. L. Hill, Thermodynamics of Small Systems, Dover, New York, 1994 Search PubMed.
- T. L. Hill, Nanoletters, 2001, 1, 273 CrossRef CAS.
- M. Temkin, Acta Physicochemica USSR, 1938, 8, 141 Search PubMed.
- C. A. Ward and J. Wu, J. Phys. Chem. B, 2007, 111, 3685 CrossRef CAS.
- D.Yu. Murzin and V.N. Parmon, Quantification of cluster size effect (structure sensitivity) in heterogeneous catalysis, Catalysis-Special Periodical Reports, RSC, vol. 23 (in press).
- M. Boudart, Kinetics of Chemical Processes, Prentice-Hall, Englewood Cliffs, NJ, 1968 Search PubMed.
- M. I. Temkin, Adv. Catal., 28, 173 Search PubMed.
- D. Yu. Murzin and I. L. Simakova, Kinet. Catal., 2010, 51, 828 CrossRef.
- D. Yu. Murzin, J. Catal., 2010, 276, 85 CrossRef CAS.
- S. Siffert, D. Yu. Murzin and F. Garin, Appl. Catal., A, 1999, 178, 85 CrossRef CAS.
- E.-J. Shin and M. A. Keane, Appl. Catal., B, 1998, 18, 241 CrossRef.
- A. Frennet, G. Lienard, A. Crucq and L. Degols, J. Catal., 1978, 53, 150 CrossRef CAS.
- A. Frennet, Catal. Today, 1992, 12, 131 CrossRef CAS.
- M. I. Cabrera and R. J. Grau, J. Mol. Catal. A, 2008, 287, 23.
- M. I. Cabrera and R. J. Grau, J. Mol. Catal. A: Chem., 2006, 260, 269 CrossRef CAS.
- H. Bernas, A. Taskinen, J. Wärnå and D. Yu. Murzin, J. Mol. Catal. A: Chem., 2009, 306, 33 CrossRef CAS.
- R. J. Berger, E. H. Stitt, G. B. Marin, F. Kapteijn and J. A. Moulijn, CATTECH, 2001, 5, 36 CrossRef.
- D. Yu. Murzin and E. Toukoniitty, React. Kinet. Catal. Lett., 2007, 90, 19 CrossRef CAS.
- B. Toukoniitty, B. Sevcikova, P. Mäki-Arvela, J. Wärnå, T. Salmi and D. Yu. Murzin, J. Catal., 2003, 213, 7 CrossRef.
- J.-P. Mikkola, T. Salmi and R. Sjöholm, J. Chem. Technol. Biotechnol., 1999, 74, 655 CrossRef CAS.
- D. Yu. Murzin, Recent Research Developments in Catalysis (Research Signpost), 2003, 2, 149 Search PubMed.
- D. Yu. Murzin, React. Kinet. Catal. Lett., 2007, 91, 37 CrossRef CAS.
- D. Yu. Murzin, Langmuir, 2010, 26, 4854 CrossRef CAS.
- M. Z. Lazman and G. S. Yablonsky, Adv. Chem. Eng., 2008, 34, 47 Search PubMed.
- G. S. Yablonsky and M. Z. Lazman, Stud. Surf. Sci. Catal., 1997, 109, 371 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |