The effect of multiwalled carbon nanotubes and activated carbon on the morphology and photocatalytic activity of TiO2/C hybrid materials
Received
8th November 2010
, Accepted 11th January 2011
First published on 15th February 2011
Abstract
TiO2
nanoparticles supported on activated carbon (AC) and multiwalled carbon nanotubes (MWCNTs) were prepared by the sol–gel method using ultrasonic irradiation. All the prepared samples were calcined at different temperatures and characterized by X-ray diffraction, nitrogen adsorption/desorption isotherms, scanning electron microscopy, transmission electron microscopy and diffuse reflectance spectroscopy. The effects of size and type of carbon on the morphology and photocatalytic activity of the resulting hybrid materials were investigated. The results showed that a decrease in the size of the carbon led to a decrease in size of TiO2 nanoparticles. Both AC and MWCNTs retarded the transformation of anatase to rutile and also prevented the agglomeration of TiO2 particles. The photocatalytic activity of the materials was elucidated using the photo oxidation of Acid Blue 92 under UV illumination. Chemical oxygen demand of the dye solution was measured at regular intervals and gave a good idea about the mineralization of the dye.
Introduction
Recently, the photocatalytic degradation of organic pollutants on a TiO2 surface has become an active area of research due to the major issue of pollution. This application is based on the semiconducting properties of TiO2.1 UV irradiation of a semiconductor can cause photoactivation of TiO2, generating electron/hole couples with strong redox properties for various environmental applications. The photocatalytic efficiency of TiO2 powder depends heavily on its microstructure and physical properties, which can be modified, while preparation is underway.2 Several attempts have been made to increase the photo degradation efficiency of TiO2 on alumina, zeolite, and activated carbon.3,4 Activated porous carbon (AC) with high adsorption capacity has been reported to improve the photocatalytic efficiency of TiO2.5,6 Additionally, several works proposed that the photocatalytic efficiency of TiO2 may depend on the surface area and TiO2 sintering temperature7 but the effect of the sintering temperature on photocatalytic activity of TiO2 outweighs the effect of surface area.8 The larger surface area probably provides the higher photoabsorption, which would increase the photocatalytic efficiency of the photocatalysts. Nevertheless, the TiO2 photocatalyst usually has a low specific surface area because of the crystalline grain growth, which occurs during calcination. Accordingly, improving the antisintering properties of TiO2 is important in increasing the photocatalytic efficiency of TiO2. However, only a few investigations have been done on improving the antisintering properties of TiO2 photocatalysts.4,9,10 Multiwalled carbon nanotubes (MWCNTs) could be considered as a good support for photocatalyst materials due to their high mechanical,11 chemical stabilities,12 and mesoporous character, which favors the diffusion of reacting species. On the other hand, dispersion of TiO2 on the MWCNT surface could create many active sites for the photocatalytic degradation. Different techniques have already been employed to deposit on the carbon support.13,14 In this study, TiO2/AC and TiO2/MWCNT hybrid materials were prepared by the sol–gel method using ultrasonic irradiation. Structural, physicochemical properties and the photocatalytic characteristics of the hybrid materials were elucidated by X-ray diffraction (XRD), nitrogen adsorption–desorption isotherms (BET), scanning electron microscopy (SEM), and diffuse reflectance spectroscopy (DRS) techniques. The photocatalytic activity of the synthesized hybrid materials was evaluated by degradation of Acid Blue 92 (AB92) under UV illumination. The chemical oxygen demand (COD) was also determined.
Experimental
Reagents
Tetraisopropylorthotitanate (Tipt, Ti (OCH(CH3)2)4), ethanol, diethanolamine and commercially available AC in two different meshes (mesh 100 (AC1) and mesh 35–50 (AC2)) were purchased from Merck. AB92 (MW = 695.58 g mol−1, C.I. 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 390) was provided by Iran Color Research Center, for which chemical structure is shown in Fig. 1. Raw MWCNTs (purity > 95%, diameter 10–15 nm, length of 30 μm, surface area 198 m2 g–1) were purchased from Aldrich. The acid treatment of MWCNTs was achieved by the procedure explained elsewhere.15
390) was provided by Iran Color Research Center, for which chemical structure is shown in Fig. 1. Raw MWCNTs (purity > 95%, diameter 10–15 nm, length of 30 μm, surface area 198 m2 g–1) were purchased from Aldrich. The acid treatment of MWCNTs was achieved by the procedure explained elsewhere.15
 |
| | Fig. 1 The structure of AB92 (C.I. 13390). | |
Characterization
XRD patterns were recorded on a D4 X-ray diffractometer using Cu Kα radiation with a wavelength of 0.15406 nm as the X-ray source. The average crystalline size of TiO2 nanoparticles was determined according to the Scherrer equation using the full width at half maximum data of each phase after correcting the instrumental broadening.16,17 The specific surface area of TiO2 nanoparticles was calculated from the nitrogen adsorption/desorption isotherms at 77 K, using a Belsorp apparatus. All samples were degassed under N2 for 4 h at 100 °C prior to measurement. The particle size and morphology of TiO2 particles were characterized using SEM (Philips XN 30). DRS of samples were performed using a GBC Cintra 40 instrument with BaSO4.
Photocatalytic experiments
Irradiation experiments were carried out in a pyrex reactor where the UV source (the UV source was provided by OSRAM 125 W high-pressure mercury lamp) was surrounded by a circulating water jacket to maintain constant temperature.18
A solution with the desired dye concentration and catalysts was fed into the reactor and the pH was adjusted using phosphate buffer. The suspension was magnetically stirred for 15 min and then the lamp was switched on to initiate the photocatalytic reaction. Samples (2 ml) were withdrawn for UV-Visible analysis at regular time intervals and centrifuged for 10 min at a rate of 10000 rpm. The dye concentration in each degraded sample was determined by a spectrophotometer (GBC Cintra 40) at λ = 574 nm.
Results and discussion
Characterization of the hybrid materials
XRD analysis.
The XRD patterns of samples are shown in Fig. 2. Characteristics of the synthesized catalysts are listed in Table 1. The TiO2 crystalline size of the samples was determined from half the width of the peaks according to Scherrer's equation. The anatase fraction (XA) of crystalline TiO2 can be calculated using the equation mentioned by Hu et al.19 As shown in Table 1 under the same calcination temperature (500 °C), the crystalline size of TiO2 in TiO2/AC is smaller than that of the pure TiO2 particles (TiO2/AC1 (12 nm) and TiO2/AC2 (15 nm)). The anatase to rutile ratio showed that the phase transforming temperature in the TiO2/AC hybrid materials is higher than that in pure TiO2. This could be attributed to the AC matrix with high surface area, which retarded the anatase to rutile phase transformation that was a result of its large interfacial energy, which creates the anticalcination effects in the AC matrix.20 By reduction of carbon active size, a decrease in the TiO2 particle size was observed. The XRD patterns of TiO2/MWCNTs and MWCNTs are demonstrated in Fig. 2b. It is obvious that MWCNTs have two characteristic peaks at positions: 26.0 and 43.4, in comparison to AC. The main peak of MWCNTs at 26.0 can overlap with the main peak of anatase TiO2 crystalline at 25.4.21 Hence, identification of the hybrid materials is difficult. Notably, the crystalline size of hybrid materials decreases gradually from 30 nm (pure TiO2) to 7 nm in TiO2/MWCNTs, which may be due to the interaction between the hydroxyl group in the titania precursor and the chemical groups on the surface defects of acid-pretreated MWCNTs,22 so MWCNTs act as a support and heat sink during heat treatments, thus stabilizing small particle sizes.23 The defects of MWCNTs may be regarded as sites of grain growth for TiO2 crystallites in the TiO2/MWCNTs. Therefore, increasing MWCNTs in TiO2 sol suppresses the agglomeration of TiO2 particles in the sintering procedure and reduces the size of TiO2 particles.24
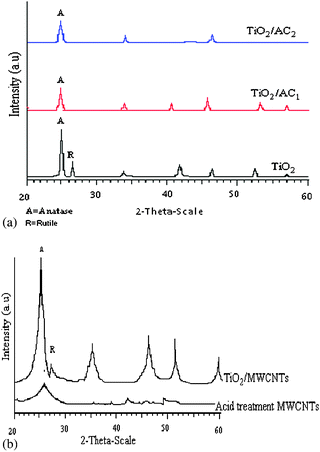 |
| | Fig. 2
XRD patterns of (a) TiO2, TiO2/AC1 and TiO2/AC2 calcined at 500 °C; (b) acid treated MWCNT and TiO2/MWCNTs calcined at 500 °C. | |
Table 1 Characteristics of the hybrid materials calcined at 500 °C
| Samples |
Aa/Rb ratio |
Crystallite size/nm |
Energy of band gap/ev |
S
BET/m2 g−1 |
Total pore volume/cm3 g−1 |
|
Anatase.
Rutile.
|
|
TiO2 |
70∶30 |
30 |
3.2 |
14.88 |
0.033 |
|
TiO2/AC1 |
75∶25 |
12 |
2.48 |
248.6 |
0.2511 |
|
TiO2/AC2 |
80∶20 |
15 |
2.75 |
206 |
0.2191 |
|
TiO2/MWCNTs |
100∶0 |
7 |
2.07 |
198.6 |
0.3814 |
BET surface area.
According to Table 1, the surface area and pore volume of the catalysts increase by the addition of AC and MWCNTs to TiO2. Generally, as the surface area of the materials is increased, the photocatalytic activity may increase. In addition, as the particle size decreases, the number of active surface sites may increase as well. TiO2/ACs have high surface area and therefore their photocatalytic activity may be expected to be higher than that of TiO2/MWCNTs. In contrast, TiO2/MWCNTs have higher photocatalytic activity than TiO2 and TiO2/AC. This phenomenon is consistent with the SEM images of hybrid materials where the MWCNT surface is completely coated by TiO2 nanoparticles. The same amount of TiO2/MWCNTs shows higher photocatalytic activity compared to TiO2/AC because of its unique structure, which causes a homogeneous distribution of TiO2 on the MWCNT surface. However, in TiO2/AC both TiO2 and AC particles are on the surface and AC particles are not active for degradation and only act as an absorbent.
TEM and SEM analysis.
The samples were investigated by both TEM and SEM.
A common characteristic of pure TiO2 observed in all hybrid materials (Fig. 3a) is a good dispersion of TiO2 nanoparticles on the surface of carbon. Two magnifications of TiO2/AC1 are shown in Fig. 3b and c. The titanium precursor may interact with the defect sites on the MWCNT surface causing TiO2 to disperse on the MWCNT surface in the calcination stage.22 The SEM image of acid treated MWCNTs is shown in Fig. 4a. Their diameter was measured to be about 28 nm. However, by increasing MWCNTs in the sol formation stage and calcination of TiO2/MWCNTs at 350 °C, very small particles are produced close to MWCNTs.
 |
| | Fig. 3
SEM images of the (a) pure TiO2 (1 μm), (b) TiO2/AC1 (10 μm), (c) TiO2/AC1 particles with high magnification (1 μm). | |
 |
| | Fig. 4
SEM images of the (a) acid treated MWCNTs (1 μm), and TiO2/MWCNTs calcined at various temperatures (the scale bars in figures represent 500 nm). | |
When the calcined temperature increased up to 450 °C, gemmating of TiO2 particles was observed on the MWCNT surface. Finally, when the calcined temperature was raised to 500 °C, homogenous TiO2/MWCNTs with 92 nm diameter were produced. As a result, MWCNTs were coated with a homogeneous layer of TiO2 with a diameter of about 64 nm.
TEM images of CNT–TiO2 samples are presented in Fig. 5. The part of CNTs covered by TiO2 shows a complete covering of TiO2. The nanosized TiO2 supported on the CNTs is observed to be homogeneously and densely spread on the surfaces of the CNTs.
DRS analysis.
The band gap is determined by the linear extrapolation of the steep part of the UV-Vis light transmission (Fig. 6) toward the base line. A larger band gap corresponds to more powerful redox ability.25 A characteristic band for titanium dioxide appeared at about 400 nm. This absorption is associated to the O2− → Ti4+ charge-transfer, related to electron excitation from the valence band to the conduction band. Observable shifts of the absorbance edge to the visible light region are detected for the C–TiO2. This shift corresponds to a decrease in the band gap energy (Table 1).
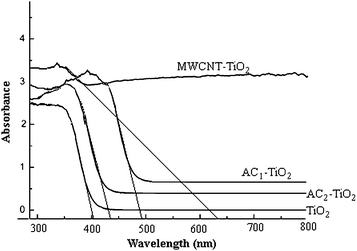 |
| | Fig. 6
DRS
spectra of hybrid materials calcinated at 500 °C. | |
Photocatalytic degradation of AB92.
Experimental results indicate that the destruction rates of photocatalytic oxidation of various organic contaminates over illuminated TiO2 correspond to the pseudo-first-order kinetics, which is displayed in eqn (1).26,27| | 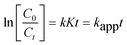 | (1) |
where C0 is the initial concentration of the reactant, Ct is the concentration of the reactant at the illumination time (t), k is the reaction rate constant, K is the adsorption coefficient, and kapp is the apparent first-order rate constant.
The apparent first-order rate constant and the photocatalytic degradation efficiency of AB92 after 120 min in the presence of TiO2/AC1, TiO2/AC2 and TiO2/MWCNTs are presented in Fig. 7a and b. Degradation rates of AB92 for TiO2/AC and TiO2/MWCNTs were faster than that for pure TiO2. In addition, the rate of degradation in the presence of TiO2/MWCNTs was two times faster than that of the TiO2/AC catalyst.
![(a) The rate constant, (b) photocatalytic degradation of AB92 in the presence of TiO2/AC1, TiO2/AC2 and TiO2/MWCNTs ([AB92] = 20 ppm, [catalysts] = 60 ppm, pH = neutral).](/image/article/2011/CY/c0cy00042f/c0cy00042f-f7.gif) |
| | Fig. 7 (a) The rate constant, (b) photocatalytic degradation of AB92 in the presence of TiO2/AC1, TiO2/AC2 and TiO2/MWCNTs ([AB92] = 20 ppm, [catalysts] = 60 ppm, pH = neutral). | |
The photocatalytic mechanism of TiO2/MWCNTs and TiO2/AC.
The photocatalytic degradation mechanism of the azo dye in the presence of catalysts can be written according to eqn (2)–(5):28–31| | | TiO2 + hν → TiO2(e− + h+) | (2) |
| | 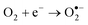 | (4) |
| | 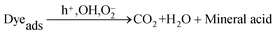 | (5) |
AC and MWCNTs facilitate the photocatalytic efficiency of TiO2 for the removal of the azo dye, as a co-adsorbent, due to their high surface area.29–31 The electrons formed by the UV irradiation can be easily transferred to the CNT–TiO2 interface without any scattering from atoms or defects. Thus, the high rate of e−/h+ pair recombination, which reduces the quantum yield of the TiO2 process, can decrease.
Effect of calcination temperature on photocatalytic activity.
The effect of calcination temperature on photocatalytic activity of hybrid materials is presented in Fig. 8a and b. TiO2/ACs and TiO2/MWCNTs showed high photocatalytic activity at 500 °C and as the temperature increased up to 600 °C, a decrease in the photocatalytic activity was observed.
![Effect of calcination temperatures on photocatalytic activity of (a) TiO2/MWCNTs, (b) TiO2/AC1 ([AB92] = 20 ppm, [catalysts] = 60 ppm, pH = neutral).](/image/article/2011/CY/c0cy00042f/c0cy00042f-f8.gif) |
| | Fig. 8 Effect of calcination temperatures on photocatalytic activity of (a) TiO2/MWCNTs, (b) TiO2/AC1 ([AB92] = 20 ppm, [catalysts] = 60 ppm, pH = neutral). | |
Nanoparticles prepared at 500 °C have the highest photocatalytic performance and by increasing the calcination temperature, the photocatalytic activity decreases. As stated before, photocatalytic activity of TiO2 nanoparticles depends on their crystalline phase and electron/hole recombination rate, which increases by augmenting the fraction of the anatase phase in the photocatalyst.32 Previous studies showed that a catalyst with good performance consists of a mixture of anatase and rutile phases with high fraction of anatase.33 The proposed reason is the transfer of electron between the crystal phases, which decreases electron/hole recombination in the electron donor phase. While, the generation rate of electron/hole is high in the rutile phase due to the lower band gap energy, the anatase phase can trap more electrons than the rutile phase and thus reduce the electron/hole recombination that leads to more holes being available. Therefore, more holes in the electron donor phase can diffuse to the surface of the crystal to take part in the radical generation step.33
Photodegradation of natural waste water.
In order to demonstrate the high photocatalytic activity of TiO2/MWCNTs, natural waste water of the textile industry was used for which the absorbent spectrum is shown in Fig. 9.
![Absorption spectra of natural waste water at different degradation times ([dye] = 20 ppm, [TiO2/MWCNTs] = 60 ppm, pH = neutral).](/image/article/2011/CY/c0cy00042f/c0cy00042f-f9.gif) |
| | Fig. 9
Absorption spectra of natural waste water at different degradation times ([dye] = 20 ppm, [TiO2/MWCNTs] = 60 ppm, pH = neutral). | |
Notably, TiO2/MWCNTs showed a decrease of absorption in all the wavelengths, which shows the high photocatalytic activity of TiO2/MWCNTs. This phenomenon was confirmed by XRD, SEM, BET and DRS results.
Estimation of COD.
COD is widely used as an effective technique to measure the organic strength of waste water. The test allows measurement of waste in terms of the total oxygen quantity required for oxidation of organic materials to CO2 and water.34 The COD of the dye solution was estimated before and after treatment. The reduction in the COD values of treated dye solution indicates that the mineralization of the dye molecules occurs along with color removal. A maximum of 86% of the degradation efficiency was obtained by TiO2/MWCNTs in the present study, which is acceptable.
Conclusions
TiO2/MWCNTs, TiO2/AC1 and TiO2/AC2 hybrid materials were prepared. All of these hybrid materials showed decrease of the crystalline size of TiO2 in comparison to pure TiO2. The most deduction was observed in TiO2/MWCNTs. As the surface area of materials is increased, the photocatalytic activity is increased as well. However, TiO2/MWCNTs that have less surface area in comparison with TiO2/ACs have higher photocatalytic activity. This phenomenon can be attributed to the morphology of TiO2/MWCNTs with a unique structure that caused a homogeneous distribution of TiO2 on the MWCNT surface (like nanotube of TiO2). Finally, the best catalyst, i.e.TiO2/MWCNTs calcined at 500 °C, was used for photodegradation of waste water of textile industry where a maximum of 86% degradation efficiency was observed in 2 h irradiation time.
Acknowledgements
The authors are grateful to Sharif University of Technology for the financial support.
References
- G. Dagan and M. Tomkiewicz, J. Phys. Chem., 1993, 97, 12651–12655 CrossRef CAS.
- J. Saien and S. Khezrianjoo, J. Hazard. Mater., 2008, 157, 269–276 CrossRef CAS.
- M. C. Lu, J. N. Chen and K. T. Chang, Chemosphere, 1999, 38, 617–627 CrossRef CAS.
- T. A. Gad-Allah, K. Fujimura, S. Kato, S. Satokawa and T. Kojima, J. Hazard. Mater., 2007, 154, 572–577.
- S. Mozia, M. Toyoda, M. Inagaki, B. Tryba and A. W. Morawski, J. Hazard. Mater., 2007, 140, 369–375 CrossRef CAS.
- W. Wang, C. G. Silva and J. L. Faria, Appl. Catal., B, 2007, 70, 470–478 CrossRef CAS.
- N. Venkatachalam, M. Palanichamy and V. Murugesan, Mater. Chem. Phys., 2007, 104, 454–459 CrossRef CAS.
- A. G. Agrios and P. Pichat, J. Photochem. Photobiol., A, 2006, 180, 130–135 CrossRef.
- V. Loddo, G. Marci, C. Martin, L. Palmisano, V. Rives and A. Sclafani, Appl. Catal., B, 1999, 20(1), 29–45 CrossRef CAS.
- B. R. Weinberger and R. B. Garber, Appl. Phys. Lett., 1995, 66(18), 2409–2411 CrossRef CAS.
- J.-P. Salvetat-Delmontte and A. Rubio, Carbon, 2000, 40, 1729–1734.
- T. Saito, K. Matsushige and K. Tanaka, Phys. B, 2002, 323, 280–283 CrossRef CAS.
- J. Li, S. Tang, L. Lu and H. C. Zeng, J. Am. Chem. Soc., 2007, 129, 9401–9409 CrossRef CAS.
- K. Byrappa, S. Dayananda, P. Sajan, B. Basavalingu, M. B. Shayan and K. Soga,
et al.
, J. Mater. Sci., 2008, 43, 2348–2355 CrossRef CAS.
- X. H. Xia, Z.-J. Jia, Y. Yu, Y. Liang, Z. Wang and L. Ma, Carbon, 2007, 45, 717–721 CrossRef CAS.
- Q. Wang, D. Yang, D. Chen, Y. Wang and Z. Jiang, J. Nanopart. Res., 2007, 9, 1087–1096 CrossRef CAS.
- N. Venkatachalam, M. Palanichamy and V. Murugesan, Mater. Chem. Phys., 2007, 104, 454–459 CrossRef CAS.
- S. Ghasemi, S. Rahimnejad, S. Rahman Setayesh, S. Rohani and M. R. Gholami, J. Hazard. Mater., 2009, 172, 1573–1578 CrossRef CAS.
- G. Hu, X. Meng, X. Feng, Y. Ding, S. Zhang and M. Yang, J. Mater. Sci., 2007, 42, 7162–7170 CrossRef CAS.
- Y. Li, S. Zhang, Q. Yu and W. Yin, Appl. Surf. Sci., 2007, 253, 9254–9258 CrossRef CAS.
- A. Jitianu, T. Cacciaguerra, R. Benoit, S. Delpeux, F. Beguin and S. Bonnamy, Carbon, 2004, 42, 1147–1151 CrossRef CAS.
- C. Y. Yen, Y. F. Lin, C. H. Hung, Y. H. Tseng, C. C. M. Ma and M. C. Chang,
et al.
, Nanotechnology, 2008, 19, 045604 CrossRef.
- N. Bouazza, M. Ouzzine, M. A. Lillo-Ro′ denas, D. Eder and A. Linares-Solano, Appl. Catal., B, 2009, 92, 377–383 CrossRef CAS.
- W. Wang, Ph. Serp, Ph. Kalck, C. Silva and J. Faria, Mater. Res. Bull., 2008, 43, 958–967 CrossRef CAS.
- Y. Ying, C. Y. Jimmy, Y. Jia-Guo, K. Yuk-Chun, C. Yan-Ke and Z. Jin-Cai,
et al.
, Appl. Catal., A, 2005, 289, 186–196 CrossRef CAS.
- Y. Li, X. Li, J. Li and J. Yin, Catal. Commun., 2005, 6, 650–655 CrossRef CAS.
- S. Ghasemi, S. Rahimnejad, S. R. Setayesh, M. Hosseini and M. R. Gholami, Prog. React. Kinet. Mech., 2009, 34, 55–76 CrossRef CAS.
- L. Lin, Y. Zhou, Y. Zhu and Y. Xie, Front. Chem. China, 2007, 2(1), 64–69 Search PubMed.
- J. Arana, J. M. Dona-Rodriguez, E. Tello Rendon, C. Garrigai Cabo, O. Gonzalez-Diaz and J. A. Herrera-Melian,
et al.
, Appl. Catal., B, 2003, 44, 153–160 CrossRef.
- X. Zhang, M. Zhou and L. Lei, Carbon, 2005, 43, 1700–1708 CrossRef CAS.
- Y. Yu, J. C. Yu, C. Y. Chan, Y. K. Che, J. C. Zhao and L. Ding,
et al.
, Appl. Catal., B, 2005, 61, 1–11 CrossRef CAS.
- J. Yu, X. Zhao and Q. Zhao, Mater. Chem. Phys., 2001, 69, 25–29 CrossRef.
- T. Wang, H. Wang, P. Xu, X. Zhao, Y. Liu and S. Chao, Thin Solid Films, 1998, 334(1–2), 103–108 CrossRef CAS.
- A. K. Subramani, K. Byrappa, S. Ananda, K. M. Lokanatha Rai and M. Yoshimura, Bull. Mater. Sci., 2007, 30, 37–41 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here. 