SPOs as new ligands in Rh(III) catalyzed enantioselective transfer hydrogenation†
Pascal M.
Castro‡
a,
Henrik
Gulyás
ab,
Jordi
Benet-Buchholz
a,
Carles
Bo
ab,
Zoraida
Freixa§
a and
Piet W. N. M.
van Leeuwen
*a
aInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: pvanleeuwen@iciq.es; Fax: +34 977-920-221
bDepartament de Química Física i Inorgànica, Universitat Rovira i Virgili, Campus Sescelades, 43007 Tarragona, Spain
First published on 1st March 2011
Abstract
The self-assembly of Secondary Phosphine Oxides (SPOs) into anionic bidentate chelates was used to construct unique systems for metal catalyzed transfer hydrogenation of ketones in isopropanol. Chelating bidentate or tridentate ligands were formed by assembly of secondary phosphine oxides through hydrogen bonding in the presence of rhodium trichloride as demonstrated by means of NMR spectroscopy and X-ray diffraction. When a chiral version of an SPO was used in asymmetric transfer hydrogenation of isopropanol and acetophenone, an enantiomeric excess of 89% was achieved. The presence of at least two ligands in the catalytically active species was confirmed by a positive non-linear effect. DFT calculations were applied to characterize several intermediates for the isopropanol dehydrogenation to produce a rhodium hydride complex and acetone. A transition state for the hydrogen-transfer was fully characterized, which revealed that the process occurs via a concerted outer-sphere mechanism.
Introduction
Secondary phosphine oxides (SPOs) act as ligands in their trivalent phosphinous acid tautomeric form (Scheme 1).1 Their metal-to-phosphorus bond is comparable in strength to that of phosphines and they are encountered as monodentate ligands, but more often as bidentate and tridentate ligands towards transition metals connected via their oxygen atoms by protons or hard metals.2 In particular, two coordinated SPOs can self-assemble after loss of one proton into a mono-anionic bidentate ligand held together strongly by an intramolecular hydrogen bond (Scheme 1), stable even in protic solvents.3–5 Hydrogen bonds are a recurring motif for the self-assembly of bidentate ligands for catalysis,5b,c but most of the hydrogen-bonded ligands will be ruptured by protic solvents, which makes these catalysts unsuitable for use in hydrogenation or hydrogen transfer reactions in such solvents. SPO complexes have been used in several catalytic reactions in protic solvents,6–8 including hydrolysis reactions,6 platinum(II) catalyzed hydroformylation of alkenes7a,b and hydrogenation of aldehydes.7d DFT calculations by Ustynyuk et al. indicated that the asymmetry of the O–H⋯O played the role of a ligand switch in these hydroformylation catalysts.7g Most likely in the latter reactions platinum does not change its valency and a heterolytic activation of hydrogen occurs. One of us proposed that the metal–SPO fragment may play a crucial role in the heterolytic splitting of H2 as it seems to be an ideal setup for this (Scheme 1),7b–e similar to cyclopentadienone-ligated ruthenium complexes as described by Casey et al.9 for Shvo's catalyst.10 Shvo's catalyst is a renowned H-transfer catalyst following a mechanism similar to that of the Noyori catalyst, akin to the heterolytic cleavage of dihydrogen.11 Mastrorilli and co-workers recently showed the activity of a diplatinum(I)–phosphido–SPO system in which H2 adds reversibly onto the Pt2–P of the phosphido fragment rather than on the SPO O-atom.12a The activity as H-transfer catalysts in combination with the supramolecular nature of SPO ligands12b,c,13 has not been exploited so far. We therefore started an investigation in the H-transfer activity of SPO complexes and particularly rhodium complexes were found to yield active and enantioselective catalysts. DFT calculations support the concerted outersphere transfer of a proton and a hydride as the mechanism in SPO–Rh H-transfer catalysts. | ||
| Scheme 1 SPOs tautomeric equilibrium, chelate formation, and heterolytic cleavage of H2. | ||
Experimental section
General procedures
All manipulations were performed under an inert atmosphere using standard Schlenk and glovebox techniques. All solvents were dried with an SPS of IT-Inc except isopropanol which was either dried over activated molecular sieves or distilled from Mg/I2. Water content was checked to be below 20 ppm by titration on a Karl Fisher titration unit Metrohm 831 KF. All reagents were purchased from commercial sources and used as received. Cyclohexanone and acetophenone (Acros) were dried over CaH2 or used as received. (R)- and (S)-dinaphtho[2,1-d:1′,2′-f][1,3,2]dioxaphosphepine-4-oxide (2) were synthesized from (R)- and (S)-[1,1′-binaphthalene]-2,2′-diol (binol), respectively, and PCl3 according to a literature procedure.20NMR spectra were recorded by using a Teflon capped or J. Young NMR tube with a Bruker Avance 400 Ultrashield NMR spectrometer or a Bruker Avance 500 Ultrashield NMR spectrometer for the variable temperature experiments. Electrospray ionization mass spectra (ESI-MS) were recorded with a Waters LCT Premier spectrometer in dry MeOH or MeCN. Gas chromatography analyses were carried out on an Agilent Technologies 6890N/G1530N spectrometer equipped with a FID detector and a Supelco Beta Dex 120 fused silica capillary chiral column (30 m × 0.25 mm diameter × 0.25 μm film thickness).
Synthesis of R-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]phosphepine-4-oxide (3)
2,2′-Di(lithiomethyl)-1,1′-binaphthyl 2 N,N,N′,N′-tetramethylethylenediamine (10.48 g, 19.9 mmol), prepared from optically pure (R)-binol according to the literature procedures,21 was treated with hexane (70 mL). Diethylphosphoramidous dichloride (3.3 mL, 22.5 mmol), in a small amount of hexane (∼3 mL), was slowly added to the suspension with a Hamilton syringe at 0 °C external temperature (ice bath). The residual reagent from the Hamilton syringe was rinsed into the reaction mixture with hexane (5 mL + 3 mL). The reaction mixture was refluxed for 4 h. The hexane was removed in vacuum. The obtained yellow solid was extracted with toluene (3 × 50 mL). The toluene was removed from the combined extracts in vacuum, and the crude 4-diethylamino-4,5-dihydro-3H-dinaphtho(2,1-c; 1′,2′-e)phosphepine was dissolved in THF (50 mL). Aqueous HCl solution (50 mL, 6 N, 300 mmol) was added at 0 °C external temperature. The ice bath was removed, and the reaction mixture was stirred at room temperature overnight. All the volatiles were removed in vacuum. The obtained yellow solid was extracted with a mixture of toluene (50 mL) and THF (60 mL). The insoluble residue, a yellowish waxy material, was washed again with toluene (50 mL). The extracts were combined, and solvents were removed in vacuum. The crude product was purified by column chromatography ((1) silica, CH2Cl2, CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶EtOH (v/v 9/1); (2) silica, CH2Cl2∶EtOH (v/v 9/1)). The product is a white foamy solid. Overall yield: 3.00 g (46%). 1H-NMR (400 MHz, CDCl3): δ = 2.94–3.14 (m, 2H, CHH, C′HH); 3.26–3.47 (m, 2H, CHH, C′HH); 7.26 (dd, JPH = 461 Hz, JHH = 7.9 Hz, P(O)H), 7.19–7.33 (m, 4H, binaphthyl); 7.45–7.56 (pseudo q, 3H, binaphthyl); 7.66 (d, JHH = 8.2 Hz, 1H, binaphthyl); 7.92–8.03 (m, 4H, binaphthyl). 13C-NMR (400 MHz, CDCl3): δ = 33.95 (d, JPC = 63 Hz, CH2); 34.60 (d, JPC = 62 Hz, CH2); 125.84 (d, JPC = 1.5 Hz, binaphthyl CH); 126.05 (d, JPC = 1.5 Hz, binaphthyl CH); 126.54 (s, binaphthyl CH); 126.70 (s, 2C overlapped, binaphthyl CH); 126.96 (d, JPC = 1.5 Hz, binaphthyl CH); 127.45 (d, JPC = 4.4 Hz, binaphthyl CH); 127.90 (d, JPC = 8.1 Hz, binaphthyl); 128.19 (d, JPC = 3.7 Hz, binaphthyl CH); 128.31 (d, JPC = 1.5 Hz, binaphthyl CH); 128.43 (d, JPC = 1.5 Hz, binaphthyl CH); 128.66 (d, JPC = 11.0 Hz, binaphthyl); 129.08 (d, JPC = 2.9 Hz, binaphthyl CH); 129.4 (d, JPC = 1.5 Hz, binaphthyl CH); 131.94 (d, JPC = 1.5 Hz, binaphthyl); 132.37 (d, JPC = 2.2 Hz, binaphthyl); 133.0 (d, JPC ≈ 2 Hz, binaphthyl) overlapped with 130.01 (d, JPC ≈ 3 Hz, binaphthyl); 133.58 (d, JPC = 3.7 Hz, binaphthyl); 134.17 (d, JPC = 4.4 Hz, binaphthyl). 31P{1H}-NMR (162 MHz, CDCl3): δ = 45.84 (s). 31P-NMR (162 MHz, CDCl3): δ = 45.84 (dm, 1JPH = 461 Hz). HRMS (TOF ES−) calculated for C22H16OP (M−H+): 327.0939. Found: 327.0942.
∶EtOH (v/v 9/1); (2) silica, CH2Cl2∶EtOH (v/v 9/1)). The product is a white foamy solid. Overall yield: 3.00 g (46%). 1H-NMR (400 MHz, CDCl3): δ = 2.94–3.14 (m, 2H, CHH, C′HH); 3.26–3.47 (m, 2H, CHH, C′HH); 7.26 (dd, JPH = 461 Hz, JHH = 7.9 Hz, P(O)H), 7.19–7.33 (m, 4H, binaphthyl); 7.45–7.56 (pseudo q, 3H, binaphthyl); 7.66 (d, JHH = 8.2 Hz, 1H, binaphthyl); 7.92–8.03 (m, 4H, binaphthyl). 13C-NMR (400 MHz, CDCl3): δ = 33.95 (d, JPC = 63 Hz, CH2); 34.60 (d, JPC = 62 Hz, CH2); 125.84 (d, JPC = 1.5 Hz, binaphthyl CH); 126.05 (d, JPC = 1.5 Hz, binaphthyl CH); 126.54 (s, binaphthyl CH); 126.70 (s, 2C overlapped, binaphthyl CH); 126.96 (d, JPC = 1.5 Hz, binaphthyl CH); 127.45 (d, JPC = 4.4 Hz, binaphthyl CH); 127.90 (d, JPC = 8.1 Hz, binaphthyl); 128.19 (d, JPC = 3.7 Hz, binaphthyl CH); 128.31 (d, JPC = 1.5 Hz, binaphthyl CH); 128.43 (d, JPC = 1.5 Hz, binaphthyl CH); 128.66 (d, JPC = 11.0 Hz, binaphthyl); 129.08 (d, JPC = 2.9 Hz, binaphthyl CH); 129.4 (d, JPC = 1.5 Hz, binaphthyl CH); 131.94 (d, JPC = 1.5 Hz, binaphthyl); 132.37 (d, JPC = 2.2 Hz, binaphthyl); 133.0 (d, JPC ≈ 2 Hz, binaphthyl) overlapped with 130.01 (d, JPC ≈ 3 Hz, binaphthyl); 133.58 (d, JPC = 3.7 Hz, binaphthyl); 134.17 (d, JPC = 4.4 Hz, binaphthyl). 31P{1H}-NMR (162 MHz, CDCl3): δ = 45.84 (s). 31P-NMR (162 MHz, CDCl3): δ = 45.84 (dm, 1JPH = 461 Hz). HRMS (TOF ES−) calculated for C22H16OP (M−H+): 327.0939. Found: 327.0942.
Transfer hydrogenation experiments
Reactions were performed in a parallel manner on a Radleys Discovery Technologies Carousel 12 reaction station. In a typical transfer hydrogenation experiment, the corresponding metal salt and the desired amount of ligand were reacted in dry isopropanol at 80 °C during one hour under an inert atmosphere to form the catalyst precursor. After this incubation period, if necessary the temperature was adjusted to the targeted value and the desired quantities of the substrate (acetophenone or cyclohexanone) and base (potassium tert-butoxide) were added. Aliquots were taken after controlled periods of time, filtered through a silica pad and analyzed by gas-chromatography to determine conversions.Computational details
All DFT calculations were carried out using the Amsterdam Density Functional (ADF2008.01) program developed by Baerends et al.22 The BP86 functional described as a combination between local VWN exchange–correlation potential with nonlocal Becke's exchange correction and Perdew's correlation correction was used. Relativistic corrections were introduced by scalar-relativistic Zero Order Regular Approximation (ZORA). A triple-ζ plus one polarization basis set was used for all the atoms. For non-hydrogen atoms a relativistic frozen-core potential was used, including 3d for Rh, 2p for chlorine and phosphorus, and 1s for carbon and oxygen. A numerical integration parameter of 6 was employed in optimization and vibrational frequencies calculations, which were evaluated analytically. Solvent effects were introduced by using the continuous solvent model COSMO23 with standard ADF radii.Results and discussion
We report here about the use of SPOs 1 and 3 and HASPO8a2 (Fig. 1) as ancillary ligands for the rhodium(III) catalyzed transfer hydrogenation of ketones in isopropanol. | ||
| Fig. 1 Ligands 1–3. | ||
In an exploratory screening intended to find an appropriate metal precursor, various metal salts were tested using diphenyl phosphine oxide (1) as the ligand, cyclohexanone and acetophenone as the substrates and isopropanol as the hydrogen donor. In general cyclohexanone was hydrogenated faster than acetophenone, many metal SPO complexes showed activity, but rhodium(III) or rhodium(I) gave the highest activities (see ESI†). The best combination involved RhCl3·3H2O (0.5 mM) for both substrates, using a metal to ligand ratio of 1∶10 for cyclohexanone (92% conversion TOF = 1825 h−1) and 1∶3 for acetophenone (18% conversion, TOF = 353 h−1).
The nature of the species formed from the reaction of RhCl3·3H2O and 1 in a 1∶3 ratio during the incubation process was first established by 31P{1H} NMR analysis in CDCl3, which revealed two doublets at 87 ppm and 82 ppm in a 3∶2 ratio (JRh–P = 124 Hz and 119 Hz) which correlates well with the data found in the literature for μ-Cl3 bridged binuclear rhodium complexes bearing anionic Ph2POH⋯−OPPh2 chelating ligands.4c At 213 K the signal at 87 ppm decoalesced to a doublet of triplets at 84 ppm and a double doublet at 89 ppm (Fig. 2). Its structure was assigned to 4 (Scheme 2) on the basis of the 2∶1∶2 ratio and multiplicity of the three signals.
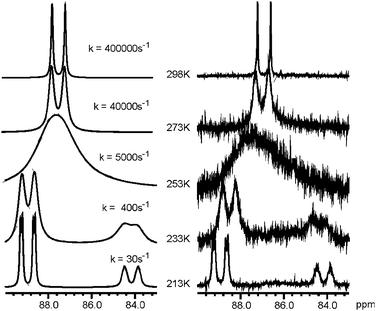 | ||
| Fig. 2 Simulated (left) and experimental (right) variable temperature 31P{1H} NMR of complex 4 (CDCl3, 201 MHz) for the signal at 87 ppm. Best-fit rate constants k are shown in the simulated spectra. | ||
 | ||
| Scheme 2 Formation of 4 by reaction of 1 with RhCl3·3H2O and generation of rhodium hydride species 5. | ||
The dynamic process is due to proton transfer and hydrogen bond exchange between the three SPOs4c (ΔH≠ = 56.9 kJ mol−1, ΔS≠ = 53.1 J mol−1 K−1, ΔG≠298 = 41.2 kJ mol−1). An electrospray mass spectrometry analysis (ESI-MS) in methanol corroborated the presence of 4. In addition a signal corresponding to [(1)4Rh2Cl5–2H] was observed (previously identified by X-ray analysis as the AsPh4+ salt).4c
The structure of 4 was elucidated unambiguously by single crystal X-ray structure analysis (Fig. 3).14 The central chloro-bridged core of the structure of 4 is similar to two symmetric structures reported in previous communications.4a,c,d The neutral complex 4 forms three intramolecular hydrogen bonds that do not interact with neighboring molecules. The two phosphinous acid protons located at the side of the Rh(III) atom linked to three phosphinous acids (P1–3) could be clearly located as Fourier peaks in the difference map. Additionally oxygen atoms O1 and O3 show larger P–O bond distances than oxygen O2, corroborating the expected positions where the protons were localized. Both hydrogen atoms are linked to the O2 atom via hydrogen bonds (distances: O1⋯O2: 2.57 Å [H1⋯O2: 1.90 Å uncorrected] and O3⋯O2: 2.49 Å [H3⋯O2: 1.65 Å uncorrected]), the three phosphorus atoms P1–3 thus forming a supramolecular analog to conventional tridentate anionic ligands.15
![Ortep-plot (thermal ellipsoids shown at 50% probability level) of 4. Non-relevant hydrogen atoms have been omitted for clarity. Selected distances (Å) Exp./[Calc.]: Rh1–P1: 2.3025(4)/[2.261]; Rh1–P2: 2.3124(5)/[2.275]; Rh1–P3: 2.2999(5)/[2.260]; Rh1–Cl1: 2.4918(4)/[2.495]; Rh1–Cl2: 2.4355(4)/[2.457]; Rh1–Cl3: 2.4855(4)/[2.474]; P1–O1: 1.5903(13)/[1.611]; P2–O2: 1.5572(13)/[1.569]; P3–O3: 1.5828(13)/[1.610]; Rh2–P4: 2.2795(5)/[2.240]; Rh2–P5: 2.2703(5)/[2.257]; Rh2–Cl1: 2.5270(5)/[2.626]; Rh2–Cl2: 2.4788(4)/[2.557]; Rh2–Cl3: 2.3650(4)/[2.443]; Rh2–Cl4: 2.3121(4)/[2.334]; P4–O4: 1.5507(16)/[1.580]; P5–O5: 1.5454(13)/[1.546].](/image/article/2011/CY/c0cy00022a/c0cy00022a-f3.gif) | ||
| Fig. 3 Ortep-plot (thermal ellipsoids shown at 50% probability level) of 4. Non-relevant hydrogen atoms have been omitted for clarity. Selected distances (Å) Exp./[Calc.]: Rh1–P1: 2.3025(4)/[2.261]; Rh1–P2: 2.3124(5)/[2.275]; Rh1–P3: 2.2999(5)/[2.260]; Rh1–Cl1: 2.4918(4)/[2.495]; Rh1–Cl2: 2.4355(4)/[2.457]; Rh1–Cl3: 2.4855(4)/[2.474]; P1–O1: 1.5903(13)/[1.611]; P2–O2: 1.5572(13)/[1.569]; P3–O3: 1.5828(13)/[1.610]; Rh2–P4: 2.2795(5)/[2.240]; Rh2–P5: 2.2703(5)/[2.257]; Rh2–Cl1: 2.5270(5)/[2.626]; Rh2–Cl2: 2.4788(4)/[2.557]; Rh2–Cl3: 2.3650(4)/[2.443]; Rh2–Cl4: 2.3121(4)/[2.334]; P4–O4: 1.5507(16)/[1.580]; P5–O5: 1.5454(13)/[1.546]. | ||
The proton corresponding to the moiety formed by the two phosphinous acids (P4 and P5) could not be easily localized, but its location was determined successfully by DFT full-geometry optimization of a model of 4, in which the phenyl substituents were replaced by hydrogen atoms. The geometry of the model indicates that this H atom is bonded to O4 and strongly hydrogen-bonded to O5 according to the computed distances (O4⋯O5: 2.457 Å [H4⋯O5: 1.356 Å]), the difference in the two H–O distances being 0.2 Å only. The strength of the other two hydrogen bonds is intermediate (O1⋯O2: 2.615 Å [H1⋯O2: 1.598 Å]; O3⋯O2: 2.620 Å [H3⋯O2: 1.607 Å]), in agreement with the X-ray structure. The trends in the Rh–P and Rh–Cl bond lengths are perfectly reproduced in the computed structure, in particular the Rh2–Cl1 bond length, which is the longest rhodium-chloride bond since this chlorine holds two anionic PO moieties in trans positions (see caption of Fig. 3).
The effect of the added base was investigated by adding 5 equivalents of potassium tert-butoxide to an isopropanol solution of 4. The solution was concentrated and the CD2Cl21H NMR spectrum of the resulting beige precipitate displayed two pseudo quadruplets around −20 ppm (JRh–H ≈ JP–H ≈ 25 Hz) which on account of NMR experiments were tentatively assigned to isomeric forms of a rhodium hydride species 5 featuring two SPOs on the rhodium nucleus bearing the hydride (Scheme 2 and ESI†).
An enantioselective version of the catalytic system was obtained using (R)-binaphthol-derived phosphorous acid 2, although it was realized that 2 might not survive the strong basic, alcoholic medium. Relatively low activities were obtained with the use of 2 (entries 2–5, Table 1), but enantiomeric excesses up to 89% were reached (entry 4, Table 1). When the catalyst concentration was lowered (entries 2, 4 vs. 3, 5 in Table 1) the ee dropped.
| Run | Ligand | [Rh]/mM | Time/min | S/Rhb | Conv. (%) | TOFc | eed (%) |
|---|---|---|---|---|---|---|---|
|
a Conditions: 1 hour incubation at 80 °C, reaction at 40 °C, [Rh]/[tBuOK] = 1∶20, [Rh]/[L] = 1∶3, total V = 10 mL.
b S = acetophenone.
c Determined by GC (TOF = molsubstrate molcatalyst−1 h−1.
d Determined by GC.
|
|||||||
| 1 | 1 | 3.3 | 240 | 1000 | 10 | 26 | <5 |
| 2 | 2 | 6.7 | 240 | 200 | 44 | 23 | 84 |
| 3 | 2 | 0.67 | 240 | 200 | 7 | 4 | 30 |
| 4 | 2 | 3.3 | 240 | 1000 | 23 | 53 | 89 |
| 5 | 2 | 0.67 | 240 | 1000 | 2 | 6 | 38 |
| 6 | 3 | 6.7 | 6 | 200 | 68 | 1364 | 8 |
| 7 | 3 | 6.7 | 240 | 200 | 79 | 42 | <5 |
| 8 | 3 | 0.67 | 6 | 200 | 19 | 376 | 14 |
| 9 | 3 | 0.67 | 30 | 200 | 97 | 387 | 13 |
| 10 | 3 | 3.3 | 6 | 1000 | 15 | 1490 | 10 |
| 11 | 3 | 3.3 | 240 | 1000 | 47 | 110 | 5 |
| 12 | 3 | 0.67 | 6 | 1000 | 10 | 971 | 26 |
| 13 | 3 | 0.67 | 240 | 1000 | 53 | 122 | 19 |
Indeed, 2 decomposes within 1 hour at 80 °C in the reaction medium as was shown by NMR and ESI-MS methods, which accounts for the low productivity. A test for a non-linear effect was undertaken by using 2 of varying degrees of enantiopurity in the catalytic reaction.16 The result was a positive non-linear effect, firmly establishing the presence of at least two chiral ligands at the active rhodium center.
To circumvent ligand decomposition, we synthesized 3, a “carbon analog” of 2 based on the dinaphthophosphepine moiety.17,18 Enantiopure R-3 (R-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]phosphepine-4-oxide) was prepared by acidic hydrolysis of the corresponding diethylamino-phosphepine. Unlike 2, in situ formed complexes of 3 provided relatively high activities, with initial TOFs reaching 1490 h−1 (entry 10, Table 1) and almost quantitative conversions (entry 9, Table 1). Unfortunately, low enantiomeric excesses were obtained.
The nature of the precatalysts formed by reaction of 3 with RhCl3·3H2O during the incubation process was inspected in a similar manner as for 1. The ESI-MS analysis displayed the presence of complexes 6 (isostructural with 4) and 7 (Scheme 3).
 | ||
| Scheme 3 Formation of 6 and 7 by reaction of 3 with RhCl3·3H2O. | ||
As mentioned in the introduction, it occurred to us that SPO complex catalyzed hydrogen transfer may involve a mechanism similar to that in Noyori and Shvo catalysts, in which an amido or an alkoxy anion participates in the hydrogen transfer.9,11 Therefore, we investigated theoretically the mechanism of isopropanol dehydrogenation to give acetone and considered two possible mechanisms: hydrogen transfer and β-H elimination. Starting from 4′, the PH2 analog of 4, we studied several ways of how isopropanol could approach the complex, and proposed first chloride/isopropanol exchange leading to species A, as shown in Scheme 4.
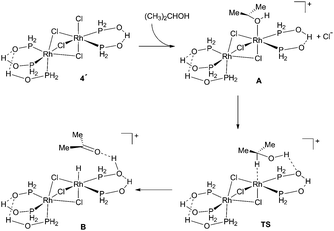 | ||
| Scheme 4 Proposed mechanism for the dehydrogenation of isopropanol. | ||
Evaluating the energy associated with this first step is not straightforward since isopropanol plays both the role of ligand and solvent, and the stability of a chloride ligand largely depends on solvation effects. By using a standard DFT GGA-based approach, the continuum solvent model method COSMO, and neglecting entropic effects, we evaluated an upper energetic limit of 20 kcal mol−1 needed to reach species A. From this point, hydrogen-transfer to the Rh–P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O fragment turned out to be facile, and a transition state (TS) structure could be fully characterized (Scheme 4, Fig. 4). Note that, in the TS, although the hydroxy proton is even more strongly hydrogen-bonded to PO, the proton has not yet been transferred. On the contrary, the C–H bond is elongated (from 1.102 to 1.479 Å) and the new Rh–hydride bond has almost formed. Recent DFT studies for the Shvo catalyst also confirmed that the hydride-proton transfer does occur in a concerted outer-sphere manner.19
O fragment turned out to be facile, and a transition state (TS) structure could be fully characterized (Scheme 4, Fig. 4). Note that, in the TS, although the hydroxy proton is even more strongly hydrogen-bonded to PO, the proton has not yet been transferred. On the contrary, the C–H bond is elongated (from 1.102 to 1.479 Å) and the new Rh–hydride bond has almost formed. Recent DFT studies for the Shvo catalyst also confirmed that the hydride-proton transfer does occur in a concerted outer-sphere manner.19
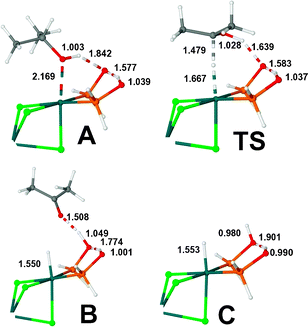 | ||
| Fig. 4 Molecular structures of A, B and C intermediates, and the transition state TS. Selected interatomic distances in Å. Only the active part of the complex is shown. | ||
The reaction proceeds towards the formation of species B in which the ketone remains hydrogen-bonded. The final step, decoordination of the ketone, leads to species C, in which the protonated oxygen maintains the chelate through the intramolecular, now weakened, hydrogen bond. Note the evolution of the chelate hydrogen-bond from 1.577 in A to 1.901 Å in C. Deprotonation of C leads to the hydride complex 5 described above.
Fig. 5 plots the computed energy for each species along the reaction coordinate. The energy barrier for the hydrogen-transfer step, from A to B, is rather low and insignificant in free energy. Formation of the hydride complex appears to be strongly exothermic. In this case, entropic factors favour the separation of B in its two constituent parts, i.e. the acetone and the hydride complex, which lie 15 kcal mol−1 below the reactants. Activation barriers of the magnitude calculated are well in accord with the rate found at molar concentrations of the substrates. Thus, complex A is an efficient catalyst for the hydrogen-transfer process following the heterolytic mechanism.
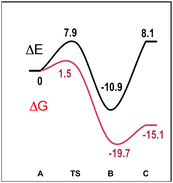 | ||
| Fig. 5 Relative energy profiles (black: electronic energy, ΔE; red: free energy, ΔG, in kcal mol−1) for the dehydrogenation of isopropanol. | ||
Complex 5 (PH2 analog) could also be obtained from A by deprotonation followed by β-H elimination in an isopropoxide complex. This reaction pathway, which involves neutral complexes, was investigated as well and several species were characterized as shown in Fig. 6. Thus, starting from the isopropoxy complex A2, the reaction proceeds to B2 by opening one chloride bridge while the isopropoxide coordination position changes from axial to equatorial, while establishing a β-H agostic interaction (Rh–β-H = 2.019). In this rearrangement one of the SPO units of the chelate ligand moves from an equatorial to an axial position. Species C2 looks very much like B2, but the agostic interaction is not present, and the SPO proton is now bonded to an SPO unit in the equatorial position. Next, in the transition state TS2 the C–H bond breaks and the new hydride–rhodium bond is formed. Note also that the former isopropoxide moiety has been transformed into acetone, which remains coordinated as in D2. The final step corresponds to decoordination of acetone and skeletal rearrangement to form again the triple chloride bridging species complex 5. It was not possible to locate transition states for opening and closing the chloride bridge. The relative energy profile in Fig. 7 shows that, although the process is exothermic, high energy barriers for both the skeletal arrangement and the β-H elimination steps are needed, the last barrier being much higher than the energy required to perform direct hydrogen transfer. According to these results, the β-H elimination reaction pathway seems less favoured than the direct hydrogen transfer assisted by the SPO ligand.
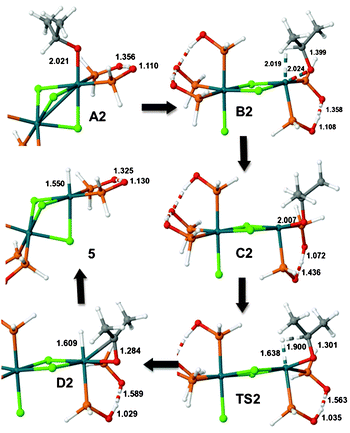 | ||
| Fig. 6 Molecular structures of A2, B2, C2, D2 and 5, and the transition state TS2. Selected interatomic distances in Å. Only the active part of the complex is shown in the left side structures. | ||
 | ||
| Fig. 7 Relative electronic energy profile (kcal mol−1) for the β-H elimination of an isopropoxide species. | ||
Conclusions
We have demonstrated that SPO ligands can be used as self-assembling chelates for catalytic reactions in protic media like transfer hydrogenation of ketones in isopropanol, thanks to the strength of the intramolecular hydrogen bond between two or three SPO units. When a chiral SPO was used, an enantiomeric excess of 89% was achieved, and the presence of at least two ligands in the catalyst, most likely in the SPO's preferred chelating mode, was confirmed by a positive non-linear effect. DFT studies revealed a plausible mechanism for the process, which occurs in a concerted outer-sphere manner.Acknowledgements
We are indebted to C. F. Roobeek for his valuable contributions. Eduardo C. Escudero-Adán, Dr Jonathan Barr, Dr Gabriel Gonzàlez and Kerman Gómez are acknowledged for technical support. The Spanish MICINN is acknowledged for “Ramón y Cajal” (ZF) and “Juan de la Cierva” (PMC) fellowships, project BAECH CTQ2005-03416/BQU, project INNOCAT CTQ2008-00683/BQU, CTQ2008-06549-CO2-02/BQU project, and Consolider Ingenio 2010 (Grant N CSD2006_0003), and the European Union for a Marie Curie Chair of Excellence Grant (PWNMvL) MEXC-CT-2005-0023600.Notes and references
- J. Chatt and B. T. Heaton, J. Chem. Soc. A, 1968, 2745 RSC.
- K. R. Dixon and A. D. Rattray, Can. J. Chem., 1971, 49, 3397.
- D. M. Roundhill, R. P. Sperline and W. B. Beaulieu, Coord. Chem. Rev., 1978, 26, 263 CrossRef CAS.
- (a) J. A. S. Duncan, D. Hedden, D. M. Roundhill, T. A. Stephenson and M. D. Walkinshaw, Angew. Chem., Int. Ed. Engl., 1982, 21, 452 CrossRef; (b) J. A. S. Duncan, T. A. Stephenson, W. B. Beaulieu and D. M. Roundhill, J. Chem. Soc., Dalton Trans., 1983, 1755 RSC; (c) J. A. S. Duncan, T. A. Stephenson, M. D. Walkinshaw, D. Heden and D. M. Roundhill, J. Chem. Soc., Dalton Trans., 1984, 801 RSC; (d) B. Patel, S. J. A. Pope and G. Reid, Polyhedron, 1998, 17, 2345 CrossRef CAS.
- (a) C. Waloch, J. Wieland, M. Keller and B. Breit, Angew. Chem., Int. Ed., 2007, 46, 3037 CrossRef CAS; (b) B. Breit, in Supramolecular Catalysis, ed. P. W. N. M van Leeuwen, Wiley-VCH Verlag, GmbH Weinheim, Germany, 2008, pp. 29–55 Search PubMed; (c) J. Meeuwissen, R. J. Detz, A. J. Sandee, B. de Bruin and J. N. H. Reek, Dalton Trans., 2010, 39, 1929 RSC.
- (a) A. J. Fanelli, G. M. Blank and F. C. Rauch, (to American Cyanamid Co., USA) U.S. Patent, 1974, US 3801639Chem. Abstr., 1974, 80, 45478 Search PubMed; (b) X.-B. Jiang, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Org. Chem., 2004, 69, 2327–2331 CrossRef CAS.
- (a) P. W. N. M. van Leeuwen and C. F. Roobeek, Eur. Pat. Appl., EP 82576, 1983Chem. Abstr., 1983, 99, 121813 Search PubMed; (b) P. W. N. M. van Leeuwen, C. F. Roobeek, R. L. Wife and J. H. G. Frijns, J. Chem. Soc., Chem. Commun., 1986, 31 RSC; (c) P. W. N. M. van Leeuwen, C. F. Roobeek, J. H. G. Frijns and A. G. Orpen, Organometallics, 1990, 9, 1211 CrossRef; (d) P. W. N. M. van Leeuwen and C. F. Roobeek, New J. Chem., 1990, 14, 487 Search PubMed; (e) P. W. N. M. van Leeuwen and C. F. Roobeek, Advances in Chemistry Series, 1992, vol. 230, Homogeneous Transition Met. Catal. React., p. 367 Search PubMed; (f) P. W. N. M. van Leeuwen, C. F. Roobeek and A. G. Orpen, Organometallics, 1990, 9, 2179 CrossRef CAS; (g) Yu. A. Ustynyuk, Yu. V. Babin, V. G. Savchenko, E. M. Myshakin and A. V. Gavrikov, Russ. Chem. Bull., 2010, 59, 686 Search PubMed.
- (a) L. Ackermann, Synthesis, 2006, 1557 CrossRef CAS; (b) M. T. Reetz and O. Bondarev, Angew. Chem., Int. Ed., 2007, 46, 4523 CrossRef CAS; (c) L. Ackermann, R. Vicente and N. Hofmann, Org. Lett., 2009, 11, 4274 CrossRef CAS; (d) T. Nemoto, M. Kanematsu, S. Tamura and Y. Hamada, Adv. Synth. Catal., 2009, 351, 1773 CrossRef CAS.
- C. P. Casey, J. B. Johnson, S. W. Singer and Q. Cui, J. Am. Chem. Soc., 2005, 127, 3100 CrossRef CAS.
- (a) Y. Blum, D. Reshef and Y. Shvo, Tetrahedron Lett., 1981, 22, 1541 CrossRef CAS; (b) R. Karvembu, R. Prabhakaran and K. Natarajan, Coord. Chem. Rev., 2005, 249, 911 CrossRef CAS; (c) C. P. Casey, T. B. Clark and I. A. Guzei, J. Am. Chem. Soc., 2007, 129, 11821 CrossRef CAS; (d) B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294 CrossRef CAS.
- (a) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS; (b) O. Pamiés and J.-E. Bäckvall, Chem.–Eur. J., 2001, 7, 5052 CrossRef CAS; (c) D. G. I. Petra, J. N. H. Reek, J.-W. Handgraaf, E.-J. Meijer, P. Dierkes, P. C. J. Kamer, J. Brussee, H. E. Schoemaker and P. W. N. M. van Leeuwen, Chem.–Eur. J., 2000, 6, 2818 CrossRef CAS; (d) Z. Chen, Y. Chen, Y. Tang and M. Lei, Dalton Trans., 2010, 39, 2036–2043 RSC.
-
(a) P. Mastrorilli, M. Latronico, V. Gallo, F. Polini, N. Re, A. Marrone, R. Gobetto and S. Ellena, J. Am. Chem. Soc., 2010, 132, 4752 CrossRef CAS;
(b) N. V. Dubrovina and A. Börner, Angew. Chem., Int. Ed., 2004, 43, 5883 CrossRef CAS;
(c) Replacing PO by PNR may lead to even a larger variety of supramolecular catalysts: F. W. Patureau, S. de Boer, M. Kuil, J. Meeuwissen, P. A. R. Breuil, M. A. Siegler, A. L. Spek, A. J. Sandee, B. de Bruin and J. N. H. Reek, J. Am. Chem. Soc., 2009, 131, 6683 Search PubMed.
- (a) M. J. Wilkinson, P. W. N. M. van Leeuwen and J. N. H. Reek, Org. Biomol. Chem., 2005, 3, 2371 RSC; (b) B. Breit, Angew. Chem., Int. Ed., 2005, 44, 6816 CrossRef CAS; (c) A. J. Sandee and J. N. H. Reek, Dalton Trans., 2006, 3385 RSC; (d) V. F. Slagt, P. W. N. M. van Leeuwen and J. N. H. Reek, Angew. Chem., Int. Ed., 2003, 42, 5619 CrossRef CAS; (e) H. Gulyas, J. Benet-Buchholz, E. C. Escudero-Adan, Z. Freixa and P. W. N. M. van Leeuwen, Chem.–Eur. J., 2007, 13, 3424 CrossRef CAS; (f) M. T. Reetz, T. Sell, A. Meiswinkel and G. Mehler, Angew. Chem., Int. Ed., 2003, 42, 790 CrossRef CAS; (g) A. Duursma, R. Hoen, J. Schuppan, R. Hulst, A. J. Minnaard and B. L. Feringa, Org. Lett., 2003, 5, 3111 CrossRef CAS.
- Crystal data for 4 at 100 K: C60H53Cl4O5P2Rh2, 1356.49 g mol−1, monoclinic, P21/c, a = 17.0585(15) Å, b = 17.1534(16) Å, c = 19.5448(17) Å, β = 90.797(2)°, V = 5718.5(9) Å3, Z = 4, ρcalcd = 1.576 Mg m−3, R1 = 0.0379 (0.0775), wR2 = 0.0842 (0.1049), for 22866 reflections with I > 2σ(I) (for 32712 reflections [Rint: 0.0463] with a total measure of 112410 reflections), goodness-of-fit on F2 = 1.074, largest diff. peak (hole) = 1.842 (−1.977) e Å−3. Taking into account the presence of a β angle close to 90° and the similarity between the a and b axes, cells with an orthorhombic and a tetragonal metric and also twin refinement were considered without success. CCDC 685489 contains the supplementary crystallographic data for this paper.
- J. C. Peters, J. D. Feldman and T. D. Tilley, J. Am. Chem. Soc., 1999, 121, 9871 CrossRef CAS.
- C. Girard and H. B. Kagan, Angew. Chem., Int. Ed., 1998, 37, 2922 CrossRef.
- S. Gladiali, A. Dore, D. Fabbri, O. De Lucchi and M. Manassero, Tetrahedron: Asymmetry, 1994, 5, 511 CrossRef CAS.
- D. Regnat, (to Hoechst A.-G., Germany), Eur. Pat. Appl., 1996, EP 703240Chem. Abstr., 1996, 125, 11132 Search PubMed.
- (a) A. Comas-Vives, G. Ujaque and A. Lledós, THEOCHEM, 2009, 903, 123 CrossRef CAS; (b) A. Comas-Vives, G. Ujaque and A. Lledós, Organometallics, 2007, 26, 4135 CrossRef CAS.
- T. C. H. Lam, W.-L. Mak, W.-L. Wong, H.-L. Kwong, H. H. Y. Sung, S. M. F. Lo, I. D. Williams and W.-H. Leung, Organometallics, 2004, 23, 1247 CrossRef CAS.
- H. Klein, R. Jackstell, K.-D. Wiese, C. Borgmann and M. Beller, Angew. Chem., Int. Ed., 2001, 40, 3408–3411 CrossRef CAS.
- (a) G. T. Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS; (b) www.scm.com, and references cited therein.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC reference number 685489. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cy00022a |
| ‡ Present address: Borealis Polymers Oy, Innovation Centre Porvoo, P.O. Box 330, FI-06101 Porvoo, Finland. |
| § Present address: Ikerbasque Research Professor, Departamento de Quimica Aplicada Facultad de Ciencias Quimicas Apdo. 1072, 20080 San Sebastian, Spain. |
| This journal is © The Royal Society of Chemistry 2011 |