Activity-based probes: discovering new biology and new drug targets
William P.
Heal
a,
T. H. Tam
Dang
a and
Edward W.
Tate
*ab
aDepartment of Chemistry, South Kensington Campus, Imperial College, London, SW7 2AZ, UK. E-mail: e.tate@imperial.ac.uk; Fax: +44 (0)20 75941139; Tel: +44 (0)20 75943752
bInstitute of Chemical Biology, South Kensington Campus, Imperial College, London, SW7 2AZ, UK
First published on 1st October 2010
Abstract
The development and application of chemical technologies enabling direct analysis of enzyme activity in living systems has undergone explosive growth in recent years. Activity-based protein profiling (ABPP) is a key constituent of this broad field, and is among the most powerful and mature chemical proteomic technologies. This tutorial review introduces the essential features of ABPP and the design and application of activity-based probes (ABPs) from drug target elucidation and in vivo visualisation of enzyme activity to comprehensive profiling of the catalytic content of living systems, and the discovery of new biological pathways.
 William P. Heal | William Heal received a BSc with honours in chemistry in 1998, and an MSc in chemical process R&D the following year from the University of Liverpool (UK). He stayed in Liverpool to complete a PhD in 2003, studying asymmetric synthesis under the supervision of Professor Stan Roberts, followed by postdoctoral research in medicinal chemistry (prion disease) at the University of Sheffield (UK). He is presently at Imperial College London (UK), undertaking postdoctoral research in the Tate group in the field of chemical biology. He is particularly interested in the design and synthesis of chemical tools for use in studying biological systems. |
 T. H. Tam Dang | Tam Dang received a MS in organic chemistry in 2007 from the University of Rostock, Germany, performing her thesis research in the lab of Prof. Peter Langer. She is currently completing her PhD thesis in the group of Dr Ed Tate in the Department of Chemistry, Imperial College London, where she has developed and applied activity-based probes (ABPs) in the pathogenic bacterium Clostridium difficile to track down new proteases and potential drug targets. Outside the lab, Tam enjoys playing the guitar, art, reading and cooking. |
 Edward W. Tate | Dr Tate received his PhD in the group of Prof. Steve Ley FRS at the University of Cambridge, and then worked with Prof. Sam Zard at EcolePolytechnique (Paris) as an 1851 Research Fellow. Following postdoctoral research in molecular microbiology at the Pasteur Institute in Paris and in chemical biology at Imperial College London, he was awarded a BBSRC David Phillips Research Fellowship in 2006, and in 2010 he was appointed Senior Lecturer in Chemical Biology in the Department of Chemistry, Imperial College London. His research group currently comprises over 20 researchers engaged in multiple aspects of the design and application of chemical approaches to understanding living systems, with an emphasis on the roles of protein modification in disease. |
Introduction
As society moves into the post-genomic era scientists face an extraordinary array of exciting challenges to decipher the emergent properties of the genome at the molecular level. Among the most pressing of these is the need to identify and characterise the catalytic activity of proteins and their complexes that serve to coordinate and control all processes in the cell. A deep spatiotemporal and quantitative understanding of enzyme activity, from cell through organ to organism, is an essential step towards a systems-level model of life, and will be a powerful tool in the increasingly complex search for viable drug targets across all diseases.Proteomics,1 the quantitative analysis of proteins present in a sample (e.g. a protein complex, a cell or an organ) has enabled many key advances in our understanding of living systems. However, to move this revolutionary technology beyond cataloguing of gene products and into the dissection of the behaviour of living systems we require ways to educe the dynamic properties of proteins that are not directly encoded in the genome. The universal identification, quantification and manipulation of emergent features such as catalytic activity, post-translational modification (PTM, Table 1), and protein complex formation may fairly be described as a ‘grand challenge’ in contemporary science. The application of chemistry to this challenge is proving particularly powerful and has given rise to a vibrant sub-field broadly termed chemical proteomics. In this review we outline the specific challenges posed by measuring catalytic activity in complex living systems, discuss the key role played by chemistry in this rapidly-developing area, and highlight important examples of how chemical tools can been designed and applied.
Catalomics: a new frontier in biology
Quantitative analysis of the catalytic content of a system has been dubbed catalomics.2 The ultimate objective of this emerging field is to provide the tools to identify and characterise all types of enzymatic activity across space (e.g. by subcellular localisation or by regions of an organ) and time (e.g. during a signalling cascade, or the development of cancer) in systems from single cells up to whole organisms. Whilst the focus of the present review is on proteins, it is worth noting that the scope of catalomics includes the ribozymes and nucleic acid–protein complexes that play primary roles in many cells.3 In this context, the process of profiling proteins by their catalytic activity is commonly termed Activity-Based Protein Profiling or Activity-Based Proteome Profiling, abbreviated to ABPP.4Catalomics presents some fascinating challenges in chemical biology (Fig. 1). The in vivo activity of enzymes is rarely a simple function of their abundance; it also depends on regulation by substrate co-localisation, post-translational modification, allosteric control, and the co-regulation of endogenous inhibitors. This is perhaps most evident in signalling cascades, where the activity of downstream effectors may be controlled on a sub-second timescale by the activity of upstream regulatory enzymes, which are in turn allosterically activated by receptor complexes. Another important example is the regulated in vivo inhibition of many proteases that is essential to prevent uncontrolled protein cleavage and degradation. These proteases are expressed as an inactive zymogen being rendered catalytically active ‘on demand’ by post-translational modification or allosteric activation.5 This highly emergent in vivo behaviour implies that cell-free experiments with recombinant enzymes can offer only limited, even misleading, information about the biological role of an enzyme. The implications for both basic biology and drug discovery are evident, as is the need for analytical tools that can access the chemistry of enzymes in living systems.
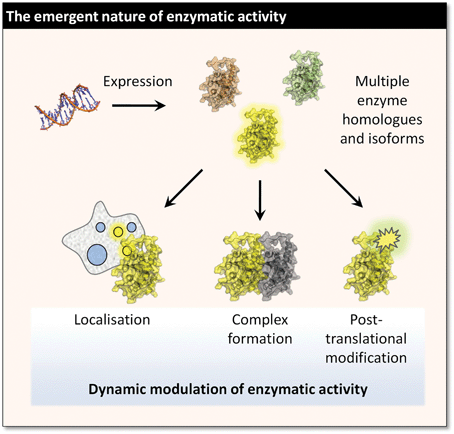 | ||
| Fig. 1 The functional catalytic properties of an enzyme in living cells and in vivo are typically modulated by a wide range of phenomena. | ||
The central role of chemistry in catalomics
Standard genomic and proteomic technologies are very blunt tools for the analysis of catalytic behaviour. The indirect and sometimes counterintuitive coupling of transcriptional activation to gene product activity is compounded by a profound mismatch in timescale between catalysis and the much slower process of gene expression. Furthermore, due to dynamic mRNA processing neither the degree of gene activation nor abundance of the gene transcript necessarily translates directly to protein abundance, which as noted above often does not correspond to the localised concentration of active enzyme. The result is at best a ‘fuzzy’ picture of enzyme activity that is poorly-resolved in space and time, and at worst a misleading dataset with estimates of enzyme activity off by orders of magnitude.6The emerging solution to this challenge is to exploit the incipient in vivo chemistry of enzymes to identify and read out their activity in situ. There are two obvious approaches to achieve this end. The more straightforward is to detect the presence of an active enzyme indirectly through use of a substrate mimic that produces a signal when it is turned over; however, this provides no information on the specific identity of the enzyme or enzyme complex that is responsible for the reaction, and requires a suitable substrate of the enzyme to be known in advance. The more nuanced approach is to detect the active form of the enzyme itself, which has the potential to provide data on both activity and identity. However, this comes at potentially increased risk of disrupting the system under study, and is not always easy to implement. Antibodies, the universal workhorse of bioanalysis, are generally limited to indirect detection of an activating PTM (e.g. protein phosphorylation or proteolytic cleavage).7 Subtle structural changes accompanying allosteric activation are not amenable to antibody detection, and antibodies are generally too cumbersome for use inside cells. The interplay between PTMs and activity can also be very complex, and PTMs are another emergent feature of biological systems that present similar challenges for analysis.8,9 Genetic engineering is also of limited use in isolation since in addition to the shortcomings of timescale noted above, direct labelling of an enzyme by a recombinant tag does not usually provide information on activation and requires the target enzyme to be known in advance. In short, a universal approach to detection and identification of active enzymes requires chemical technologies that can address enzyme chemistry directly in living systems.
Activity-based probes (ABPs)
Chemical probes that directly detect the formation of a catalytically active site in an enzyme are termed ‘activity-based probes’ (ABPs), or ‘mechanism-based probes’. ABPs rely on some aspect of the catalytic mechanism of the enzyme to ensure that detection occurs only for catalytically-active species. This might include direct alkylation of a specific nucleophilic group at the catalytic site, or enzymatic conversion of the ABP into a highly reactive intermediate that then crosslinks to proximal residues. In contrast to straightforward substrate mimetics (for example fluorogenic substrates) and reversible inhibitor, ABPs remain covalently bound to the enzyme active site, and in this way impede subsequent catalytic activity in the same manner as a ‘suicide substrate’ or irreversible inhibitor. By attachment of a suitable handle (for example, a fluorescent label) to the ABP, formation of the active enzyme may be detected directly. As we will see, ABPs have a wide range of current and potential applications in catalomics and beyond; however, at least in living systems, the trade-off is potential disruption by (partial) inhibition of the target enzyme.Activity vs. affinity
Before examining some of the myriad embodiments of ABPs, it is worth noting the sometimes subtle distinction between activity- and so-called ‘affinity-based’ probes (Fig. 2). An affinity-based probe, conveniently abbreviated AfBP by the Yao group,10 achieves labelling by binding at a specific site on a protein (not necessarily an enzyme active site) followed by a non-specific covalent bond-forming event, for example photochemical cross-linking or spontaneous trapping of a nearby (non-catalytic) protein functional group. Careful design of an AfBP can result in the generation of a functional ABP; for example, a substrate mimic that binds preferentially to a well-formed active site coupled to a photo-activated linker. The distinction resides in the need for a catalytically-active enzyme: if a catalytic site residue that is essential for enzyme chemistry but non-essential for substrate recognition is inactivated (e.g. by mutation) an AfBP may be able to function as normal. However, an ABP would not, as labelling relies on the specific chemistry that the enzyme performs for its activation. AfBPs lie largely outside the scope of this review, but they can be potent tools for the identification/validation of the targets (and off-targets) of drugs and inhibitors, and for general protein-labelling.2,11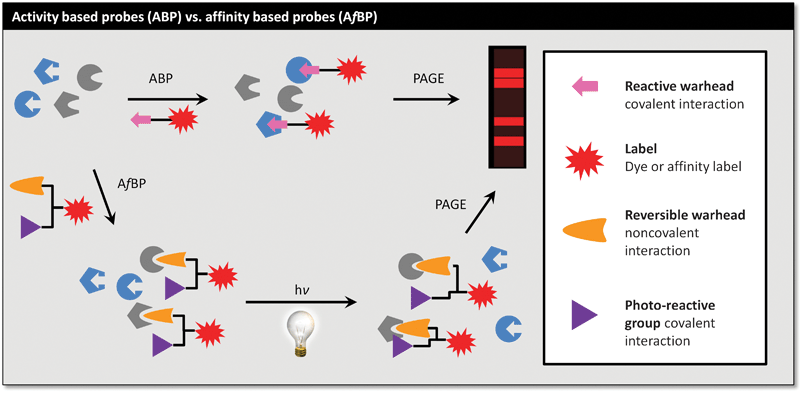 | ||
| Fig. 2 ABPs label on the basis of activation by the enzyme, whilst AfBPs label on the basis of affinity, stabilised by a non-specific cross-linking. | ||
Designing ABPs: warheads, specificity and tags
Appropriate exploitation of catalytic site chemistry is an essential feature of effective ABPs. The design of most ABPs (and AfBPs) is linked directly to the design of effective enzyme inhibitors. Indeed, the generation of most of these probes has emerged from the discovery of irreversible and reversible inhibitors that can be converted into ABPs and AfBPs, respectively, by careful molecular engineering.An ABP may often be generated directly from a given irreversible inhibitor, which is in turn frequently derived from a substrate mimetic or natural product.12,13 There are also recent instances of ABPs designed in a ‘reverse’ manner by converting a known reversible inhibitor into an irreversible ABP.14 In the case of ABPs, the response to enzyme chemistry is usually encapsulated in a chemical warhead, for example an electrophilic trap such as an epoxide or Michael acceptor. Upon activation by the enzyme the warhead reacts irreversibly to form a new covalent bond to the active site (Fig. 3). By linking enzyme chemistry to ABP activation the warhead typically defines the catalytic class of enzyme targeted enabling distinction, for example, between proteases, phosphatases or glycosidases. The reactivity of the warhead with respect to the enzyme active site depends on both affinity and innate chemical reactivity, and tuning this reactivity offers control of selectivity between enzymes. Highly reactive warheads tend to be promiscuous, whilst others can have negligible reactivity in living systems unless coupled to an element that provides increased affinity, resulting in greatly increased reaction rates for a well-defined set of targets. It is often possible to generate an ABP with either very broad or more selective labelling by including a specificity element that directs the ABP to a particular class of enzymes or to a specific enzyme within a class.
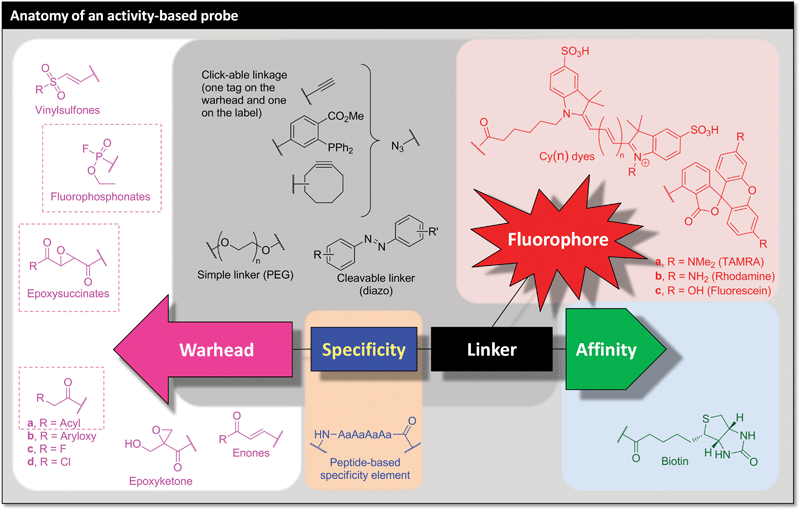 | ||
| Fig. 3 Summary of the key features of an activity-based probe (ABP), with some representative examples. | ||
The key component of an ABP that sets it apart from a common irreversible inhibitor is the presence of a tag or label (Fig. 3). This provides the ‘handle’ with which to manipulate, visualise or quantify the active enzyme: in the act of activating and reacting with the ABP the enzyme labels itself. This handle can range from the simple attachment of a dye molecule directly to the ABP through to advanced labelling technologies that can introduce sets of labels starting from a minimal chemical ‘tag’ by exploiting bioorthogonal ligation or ‘click’ chemistry.15–17 An important caveat is that a label directly appended to the ABP may interfere with the selectivity or activity of the probe, and the use of a ‘clickable’ tag has proven a reliable way to avoid this potential pitfall in many systems.2 ABPs have even been designed to be fluorogenic, switching on their fluorescence in response to enzymatic activation and thereby encompassing certain properties of a fluorogenic substrate. Here we will use the term ‘tag’ to refer to a ‘clickable’ functional group (typically an alkyne) for indirect (‘two-step’) labelling, and ‘label’ to refer to directly labelled ABPs. This distinction between a ‘tag’ and a ‘label’ can be useful, but it is important to note that these semantics are not universally observed by researchers in the field. Recent reports have added a fourth component, a targeting element, which serves to enhance cell uptake or to deliver the ABP to a specific cell type in vivo,18,19 or chemically-cleavable linkers to facilitate selective release of targets.20,21
The modular nature of ABPs (warhead, specificity element, tag/label) lends them to modular synthetic approaches such as solid-phase (peptide) synthesis (SP(P)S). In ideal cases the three components can be varied independently within a single synthetic scheme; this enables rapid access to libraries of variants that can be screened across multiple enzyme classes and exploit orthogonal detection strategies. Click chemistry has also proven a useful tool for the rapid modular assembly of ABPs, as recently reviewed by Yao et al.7
Applying activity-based probes
In addition to the key advantages outlined above, chemical probes are readily applied across multiple target species and disease models (provided probe delivery and bioavailability are maintained), a feature they share with post-genomic technologies such as proteomics and chemical genetics. One may contrast this with systems for genetic manipulation which, in addition to the drawbacks previously noted, must also be reengineered for each organism and adapted to each target enzyme. Cross-species probe portability is especially useful in organisms that lack robust technologies for genetic engineering, including a number of very important pathogenic species; in these cases ABPs can provide fundamental biological information that is currently unobtainable by other means. As a further benefit, ABP-enabled catalomics typically detects the individual activities within homologous groups of enzymes, potentially picking up compensatory modulation of a parallel biochemical pathway in response to a stimulus (e.g. chemical inhibition, RNA interference). Some of the established and emerging applications for ABPs within the general realm of catalomics include:• De novo enzyme discovery: (re)assignment of roles for proteins. This is a key challenge since around 30–40% of gene products of the human genome (and a much larger proportion in many other organisms) currently have no known role.
• Context-independent high-throughput assays for enzyme activity (as opposed to simple expression level). Even where the substrate is unknown and a conventional enzyme activity assay would be difficult to devise, assays using ABPs can enable high-throughput screening.
• Competition assays to determine in vivo selectivity of a reversible or irreversible inhibitor across a particular enzyme class. ABPs enable selectivity to be determined across all potential off-targets, including those with uncharacterised function.
• Fluorogenic probes for enzyme labelling, localisation of enzyme activity by microscopy and whole organism imaging, quantification of enzyme activity in vivo.
• Tools for probing the protein chemistry of enzymes: these probes can also be useful in structural and biochemical studies to manipulate the active site of an enzyme without resorting to mutagenesis.
• In the second part of this review, we will look at three selected case studies that highlight some of these applications.
Case study 1: dissecting lipid signalling in living systems
Since the emergence of activity-based protein profiling in the early 1990s, the Cravatt lab (currently at the Scripps Research Institute) has made a major contribution to the development of ABPs and associated technologies. An excellent recent example of this work concerns the elucidation of the role of monoacylglycerol lipase (MAGL), an enzyme highly expressed in aggressive human cancer cells, in tumour lipogenesis.22 The modification of cellular metabolism accompanies oncogenesis, and this is achieved by regulation of certain key enzymes. The ability to profile changes in enzyme activity levels in a dynamic manner provides a means for the dissection of metabolic pathways and the discrimination of enzymes whose over-activity is required for the development of cancer. One such disrupted metabolic pathway is thought to be lipogenesis, de novo lipid biosynthesis, based on the observation that alteration of fatty acid synthase (FAS) levels relate positively to disease progression in breast cancer patients.23 However, as fatty acids are biosynthesised they are quickly sequestered by lipid stores, implying that a corresponding ‘lipolytic’ process is required, to release these fatty entities for the metabolic/signalling mechanisms of oncogenesis. Using ABPP, Cravatt was able to show that the activity of the lipolytic enzyme MAGL was significantly upregulated in several aggressive cancer cell models.To start with, a catalomic analysis of a range of aggressive and non-aggressive melanoma, ovarian and breast cancer cell lines was carried out to identify enzymes possessing significant hydrolytic activity (Fig. 4). ABPP using a fluorophosphonate (FP) ABP was carried out, as this functionality is directed towards serine hydrolases,24 which represent one of the largest and most varied groups of hydrolytic enzymes in the human proteome. With roles in proteolysis, signal transduction and lipid processing, it was hoped that hydrolases with modified activity unique to the aggressive lines would be identifiable. The FP functionality reacts to form a covalent bond with the catalytic serine nucleophile of the serine hydrolases, and a FP-biotin probe can distinguish between active and inactive forms of these enzymes. This allows monitoring of enzyme activity, even when this is not related to enzyme expression level. After treatment of the soluble and insoluble cancer cell proteome fractions with the FP-biotin probe, functionalised entities were enriched by a two-stage process of size exclusion chromatography to remove unreacted probe followed by incubation with avidin–agarose beads. Tryptic digestion of enriched enzymes directly from the beads preceded separation of this complex mixture by two-dimensional liquid chromatography on a biphasic (strong cation exchange/reverse phase) capillary column with elution over a 5-step gradient of salt concentration.
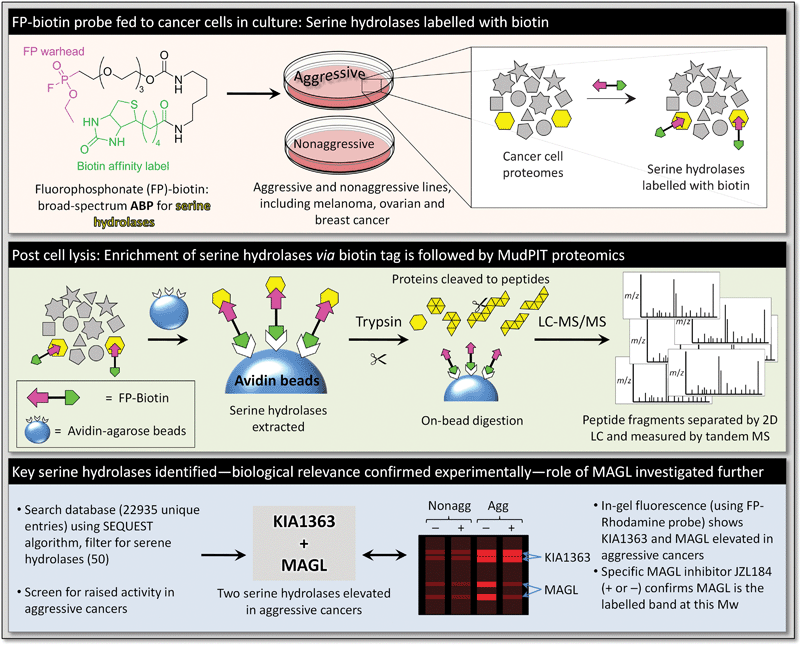 | ||
| Fig. 4 Strategy for identification of MAGL as a principle component of the metabolic pathways of aggressive cancers. | ||
Along with analysis of the LC output by ion trap tandem mass spectrometry, this system comprises the so-called multidimensional protein identification technology (MudPIT).25 The mass spectra obtained were analysed against a custom database containing over 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 unique gene products and the results filtered for serine hydrolases. Of the >50 serine hydrolases detected in this way, two (KIAA1363 and MAGL) exhibited consistently raised activity levels in aggressive lines. Whilst the authors had shown previously that KIAA1363 was involved in lipid signalling in aggressive cancer cell lines,26 little was known about the function of MAGL. Elevated MAGL activity was confirmed by monitoring the hydrolysis of the substrate C20∶4 MAG, and upon treatment with a potent and selective MAGL inhibitor (JZL18427) MAG hydrolysis was substantially reduced. The authors demonstrated that highly elevated MAG hydrolysis is an important feature of aggressive cancers that is mostly, if not completely, the result of up-regulation of MAGL activity. MAGL was also found to regulate free fatty acid (FFA) levels in aggressive cancer cells, which is not generally the case in normal cells. Cravatt went on to demonstrate that disruption of MAGL activity (by knock-down and by JZL184 treatment) reduced aggressive cancer pathogenicity, and that inducing over-expression in non-aggressive lines increased their aggressive character.
000 unique gene products and the results filtered for serine hydrolases. Of the >50 serine hydrolases detected in this way, two (KIAA1363 and MAGL) exhibited consistently raised activity levels in aggressive lines. Whilst the authors had shown previously that KIAA1363 was involved in lipid signalling in aggressive cancer cell lines,26 little was known about the function of MAGL. Elevated MAGL activity was confirmed by monitoring the hydrolysis of the substrate C20∶4 MAG, and upon treatment with a potent and selective MAGL inhibitor (JZL18427) MAG hydrolysis was substantially reduced. The authors demonstrated that highly elevated MAG hydrolysis is an important feature of aggressive cancers that is mostly, if not completely, the result of up-regulation of MAGL activity. MAGL was also found to regulate free fatty acid (FFA) levels in aggressive cancer cells, which is not generally the case in normal cells. Cravatt went on to demonstrate that disruption of MAGL activity (by knock-down and by JZL184 treatment) reduced aggressive cancer pathogenicity, and that inducing over-expression in non-aggressive lines increased their aggressive character.
This work is an elegant example of ABPs in action: starting from a black box of non-aggressive vs. aggressive phenotype, broad-spectrum serine hydrolase probes were used to identify candidate enzymes potentially involved in aggressive cancers de novo from live cells, picking up activity differences that would not have been detected at the expression level. The same probes were used to identify selective inhibitors for MAGL, and to verify modulation of its activity during target validation studies. As a direct result new insights into lipid metabolism in cancer have been gained, and a new drug target has been uncovered for aggressive cancers. In future, detection of enzyme activity using ABPs may be used as an effective biomarker for early detection of aggressive cancers, something that standard transcriptomic or proteomic analyses typically fail to discriminate.
Case study 2: caspase probes for optical imaging in vivo
Due to the obvious healthcare benefits—including early diagnosis, monitoring disease progression and assessing chemotherapeutic response—non-invasive optical imaging of diseases in whole organisms is an area of intense research. There is a pressing need for more agents that allow precise detection of individual biochemical changes in real time, in vivo, and the application of ABPs in this field has been pioneered by the group of Bogyo (based at the Stanford University School of Medicine).18Apoptosis is a programmed and tightly controlled mechanism for cell death, which in normal tissues is an important part of homoestasis and for the destruction of damaged cells. However, incorrect regulation of apoptosis has been estimated to play a role in as much as 70% of human diseases.28 Our understanding of the importance of apoptosis, a form of programmed cell death, in cancer, and of the roles played by proteases in this process, is amongst the most significant breakthroughs in cancer biology in recent years. Members of the cysteine protease family, cysteine-dependent aspartate-directed proteases, or caspases, are the principle executors of the apoptotic process.29 They are involved with activation of DNases (e.g. caspase activated DNase, or CAD), inhibition of DNA repair enzymes (such as poly (ADP-ribose) polymerase, or PARP) and digestion of structural proteins in the nucleus (e.g. lamin, which mediates interactions between chromatin and the nuclear membrane and helps maintain the shape of the nucleus). Each of these functions acts to reduce the genetic and structural integrity of the cell to facilitate its organised non-inflammatory destruction, an essential mechanism by which the body limits carcinogenesis.
Although labelled analogues of annexin V have been reported as probes for apoptosis, they suffer from slow clearance in vivo, resulting in high background.30 Alternatively, Bogyo et al. have shown that acyloxymethyl ketones (AOMK, a chemical warhead) are effective ABPs for labelling caspases in vivo, but they also display significant cross-reactivity with lysosomal cysteine protease legumain and cathepsins.31 However, by iterative optimisation of the specificity of the peptide sequence (using positional scanning libraries, obtained by solid phase peptide synthesis), it proved possible to lower the legumain response and eliminate the cathepsin cross-reactivities altogether. The probe of choice AB50-Cy5 also contains the fluorescent cyanine fluorophore Cy5 (Fig. 5). In addition, the inclusion of well-known cell penetrating peptide trans-activating transcriptional activator (Tat, from HIV-1)32 into the probe produced enhanced uptake and greatly boosted the signal in apoptotic cells and tissues, giving multifunctional probe tAB50-Cy5. The effective inclusion of a targeting element is an excellent example of the versatility possible in ABP design.
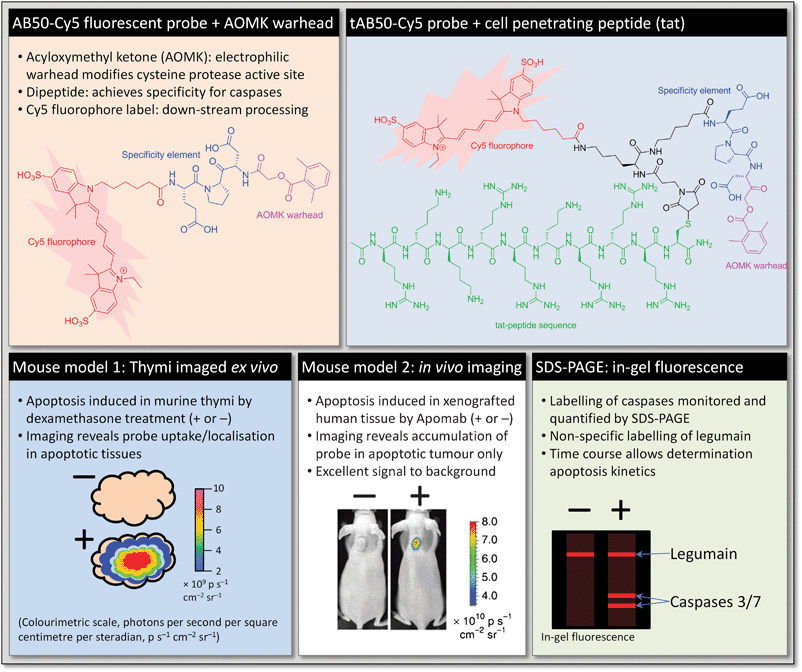
To demonstrate the efficacy of these ABPs in probing for caspases Bogyo et al. monitored apoptosis in the CD4+CD8+ thymocytes of mice treated with dexamethasone, a well-characterised in vivo system which has been studied previously with apoptosis markers such as annexin V.33 Mice were monitored over several time points after treatment with dexamethasone and intravenous injection of ABP two hours prior to removal of the thymi for imaging. After imaging, protein was extracted from the tissue and separated by SDS-PAGE, using in-gel fluorescence to visualise labelled proteins. By immunoprecipitation the authors were able to show that caspase-3 and legumain were labelled with probe. Quantification of the total caspase and legumain, and comparison with the data from intact thymi indicated that legumain activity was low and remained so after dexamethasone treatment, whereas caspase-3 was seen to become active rapidly after treatment, peaking at 12 h and dropping to background levels by 24 h. Furthermore, comparing the imaging data from the intact thymi and direct quantification showed that the overall trends are correlated and that whole-organ imaging is a sufficient readout for labelled caspases.
Whilst probing with AB50-Cy5 produced similar labelling patterns, there was a marked increase in fluorescence from intact thymi labelled with tAB50-Cy5. This was not the case in tissue in which apoptosis had not been induced, indicating that the tat peptide was facilitating uptake only in apoptotic cells. Further analysis by flow cytometry showed that, by comparison of fluorescence in apoptotic vs. non-apoptotic cells, the tAB50-Cy5 probe accumulated in the dying cells only. In a further demonstration of the specificity of these probes, intact thymi at 12 h after dexamethasone treatment showed high levels of probe accumulation relative to untreated controls, but with no caspase labelling observable by SDS-PAGE. This demonstrates the significant uptake of probe by cells as they become apoptotic, and precise labelling of caspases dependant on their level of activity.
To demonstrate the utility of these probes in non-invasive imaging, and in a model more closely related to human disease, apoptosis was studied in mice that had undergone human tumour tissue xenografts and in which apoptosis had been induced by the monoclonal antibody Apomab.34 As well as bearing more relevance to human disease, the mechanism of induction of apoptosis of Apomab is entirely distinct from that in dexamethasone treated thymocytes discussed earlier. Initial experiments mirrored those discussed earlier: following dosing with Apomab probe was administered at specific time points, and after a further 1 h whole tumours were excised and imaged ex vivo. This allowed comparison of image data and quantification of caspase labelling by SDS-PAGE. Similarly to the thymi experiments before, fluorescent signal intensity in whole tumour imaging correlated closely with the quantities of labelled caspase, with legumain labelling remaining steady over the time points studied. Peak caspase activity was found to occur 12 h after administration of Apomab. This represents a direct biochemical monitoring of the kinetics of apoptosis in vivo. To optimise clearance (and thus signal to noise), 12 h after treatment with Apomab probes were administered to mice and tumours imaged in a non-invasive manner over several time points. Interestingly, in this study it was found that the probe bearing the tat-peptide, tAB50-Cy5, whilst producing an overall stronger fluorescence signal, exhibited considerably slower clearance than AB50-Cy5. Uptake into tumours was also shown to be apoptosis specific, with SDS-PAGE confirmation ex vivo to show that caspases were labelled specifically in apoptotic tumour tissues.
Taken together, this work presents a highly elegant development of probes that allow very early non-invasive detection of apoptosis along with direct real time kinetics measurements of protease activity, and these tools will clearly be of considerable value in preclinical and possibly also clinical applications. It is also an excellent example of fine-tuning the specificity and targeting elements of an ABP to achieve a desired objective. Taking advantage of the modular nature of ABPs and the power of solid-phase synthesis technology, the authors were able to eliminate an undesired activity whilst moderating a second off-target activity to act as an internal control for labelling, and achieve superior bioavailability of their probes in vivo. A comparative study by the same lab has recently shown that fluorogenic ABPs can be superior to fluorogenic substrates for the detection of enzyme activity, particularly in an in vivo setting.35 This may seem counterintuitive since ABPs do not benefit from signal amplification due to enzyme turnover in the same way as substrates. However ABPs, which tend to be small and drug-like, benefit from better bioavailability, rapid clearance and prolonged retention at the site of action by virtue of remaining covalently bound to the target enzyme. These results in low background combined with a less diffuse signal that remains strictly dependent on enzyme activity.
Case study 3: targeting surface layer formation in C. difficile
In a recent paper from our own group we reported the development of targeted ABPs for the purpose of inhibitor development, target identification, and investigation of functional biology in a particularly challenging pathogen, Clostridium difficile.13C. difficile, a Gram positive bacterium, is a leading cause of hospital-acquired infection and is highly resistant to most common antibiotics.36 It possesses a dense semi-crystalline proteinaceous surface layer (the S-layer) that mediates host–pathogen interactions and provides great physicochemical resistance. The key structural components of the S-layer derive from proteolytic cleavage of a precursor protein called SlpA into high- and low-molecular-weight S-layer proteins (the HMW and LMW SLPs). During cell division upwards of 400 molecules of SlpA per second per cell are synthesised, translocated to the S-layer, cleaved and inserted into the matrix. This phenomenal rate of protein maturation strongly suggests both a critical need for this protein in the S-layer and a specific biological mechanism for concerted S-layer biogenesis. At the outset of our work inhibitors of S-layer biogenesis were unknown, as was the identity and cellular location of the putative protease involved in SlpA cleavage. Furthermore, the genetic tools for manipulating C. difficile are very limited in comparison to model organisms such as E. coli, making complex processes difficult to study.We decided to take a direct catalomic approach to identify the protease by its activity in vivo, and the first step therefore was to identify a suitable inhibitor as a starting point for ABP development. A live cell assay for SlpA cleavage was used to screen a small library of known protease inhibitors with diverse activities: by measuring the quantity of processed vs. full-length SlpA in the S-layer of treated C. difficile cultures we were able to identify a handful of compounds capable of weak inhibition of S-layer biogenesis. Of these, the natural product E-64 (Fig. 6) was most promising; this compound is a broad-spectrum cysteine protease inhibitor, and typically acts by covalent modification of the unique active site cysteine.37 The warhead in this case is an epoxysuccinyl moiety, and the specificity element is based on L-leucine and a guanidino motif that resembles the side chain of arginine.
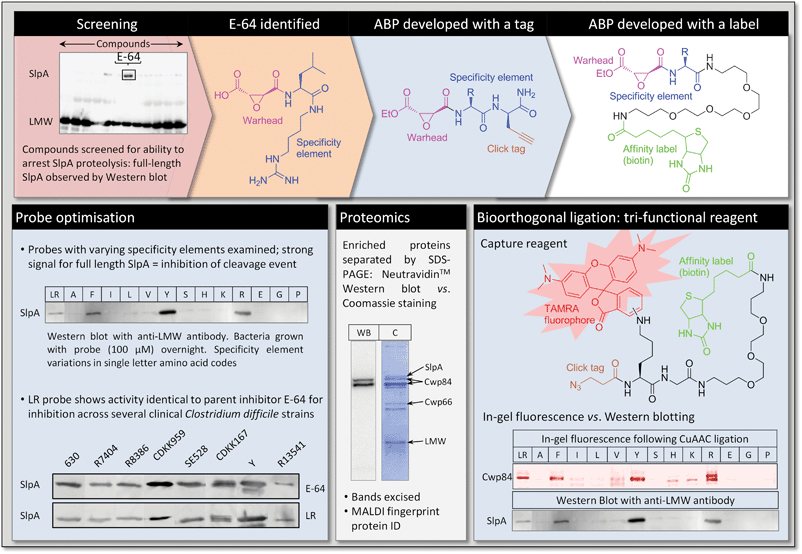 | ||
| Fig. 6 Inhibitors and ABPs used to target and identify Cwp84, the protease that mediates S-layer biogenesis in C. difficile. | ||
Taking the E-64 warhead as our starting point, we re-engineered the specificity element initially to Leu-Arg; this inhibitor recapitulated the activity of E-64, and could be accessed using an efficient solid-phase synthetic strategy. Synthesis of a wide range of analogues varying amino acids at both positions demonstrated that only the first position (Leu in E-64) has a strong influence on S-layer processing (Fig. 6). We therefore designed a first generation ABP that incorporated propargyl glycine as the second amino acid, thus introducing a tag into inhibitors for subsequent labelling by click chemistry. Amongst these ABPs we found that those incorporating Tyr, Phe and Arg exhibited greatly improved inhibition over E-64 in a cell-based assay, whilst others lost all activity (e.g. Ala, Ser, Glu). Interestingly, this specificity correlates with the consensus sequence for SlpA cleavage, strongly suggesting that the inhibitors bind to the enzyme in a manner similar to the substrate.
With a spectrum of ABPs in hand, we developed a protocol for the click labelling step; this proved challenging, as the high salt conditions used for sample preparation from C. difficile cultures interfered with the ligation reaction. Indeed, it is important to note from a practical perspective that bioorthogonal ligation can require significant optimisation when applied to the highly complex mixtures obtained from whole organisms, and there remains scope for development of more robust protocols and reagents. Exploiting a novel dual-label reagent we were able to introduce both a fluorescent dye and a biotin moiety onto target proteins (Fig. 6).38 This relatively new development in labelling chemistry provides sensitive, convenient detection and quantification (using in-gel fluorescence), and an affinity handle (biotin) for chemical proteomics. Using these two-stage ABPs, we demonstrated labelling of a protein that correlated with S-layer inhibition activity, along with a degree of background labelling due to off-target ligation. This target protein appeared to be in the surface layer of the bacterium, and we therefore switched to membrane-impermeable ABPs directly-labelled with biotin. These probes enabled very specific labelling of the target protease, and its subsequent isolation and identification by proteomics as the putative cysteine protease Cwp84. Interestingly, we also identified components of a putative processing complex that co-purified with Cwp84 and an ABP that appears to intercept multiple isoforms of the protease, suggesting a complex maturation pathway in vivo.
The work described above is one of a growing number of examples of how ABPs can assist in the dissection of a complex biological process in an organism that is decidedly difficult to manipulate using the tools of molecular biology. Faced with essentially a ‘black box’ in which only the final processed S-layer proteins were open to direct analysis, the authors sought to prove the hypothesis that the S-layer precursor is cleaved by a single specific protease at the cleavage site, as defined by proteomics studies. This is a common challenge in biology: the phenotype (in this case, S-layer assembly) is known, but the mediator (here, a protease) is unknown. The classical genetic approach would generate knockout strains of every gene (or putative protease gene) in the genome, and observe whether cleavage still occurs. However, this approach is practicable only in well-studied model organisms with elaborate tools for genetic engineering, and can be particularly difficult where multiple proteins are involved or where a process is essential, making a knockout strain non-viable. In such cases chemical inhibitors and ABPs are uniquely powerful, providing information on enzyme activity that is not available by means of genetic manipulation.
Ultimately, the ABP approach enabled us to identify a C. difficile cysteine protease (Cwp84) de novo by its catalytic activity, define its localisation in the S-layer and verify conservation of its activity across multiple C. difficile strains. Using an S-layer processing system reconstituted in E. coli we demonstrated on-target processing of SlpA by Cwp84 and showed that specificity is dictated largely by the P2 residue of the substrate, which is also likely to be the binding site for the ABPs. Furthermore, this project led to the discovery and optimisation of the first inhibitors of S-layer biogenesis, with EC50 values in the low-μM range. Finally, labour-intensive genetic manipulation of C. difficile has led to a Cwp84 knockout strain that proves unequivocally that Cwp84 is the only protease responsible for the cleavage of SlpA into the major S-layer components.39
Summary
Development of an ABP for a specific process requires as a starting point a mechanism-based inhibitor that can act irreversibly, and often this must be discovered de novo by a cell-based phenotype screening process. Efficient in vivo assays for a specific phenotype can be challenging to develop, and throughput is typically low. Common examples include Western blotting to identify an effect at the protein level (for example, failed S-layer processing), or microscopy-based screens to detect changes at the cellular level, often using fluorescent labels. It is therefore necessary to bias the screen towards inhibitors that are more likely to influence the phenotype, as in the case of S-layer biogenesis, where broad-spectrum protease inhibitors were selected. As shown above, subsequent inhibitor optimisation based on features of a known substrate can lead to more selective inhibitors, and an ABP that provides a specific readout for a particular enzyme activity in vivo. Alternatively, a broad-spectrum ABP can be used to provide a superset of potential targets, and libraries screened against this probe to identify selective inhibitors by competition. Access to ABPs with a spectrum of inhibition activity can be beneficial since this makes it possible to correlate labelling efficiency with a structure–activity relationship (SAR), providing enhanced confidence in the assignment of function. Previous studies have used Western blotting against biotin rather than fluorescence for detection of activity, but for two-step labelling this is prone to interference from excess reagents used in the bioorthogonal ligation and is non-linear, giving a relatively homogenous signal that does not correlate well with labelling efficiency. It is also worth noting that detectable levels of enzyme labelling often manifest at much lower (often >10-fold lower) concentration than an inhibition phenotype, and more effective ABPs are most easily distinguished by fluorescence.Emerging targets in catalomics
Whilst this review is intended to provide an illustrative and introductory rather than comprehensive overview of ABP design and application, we would also like to draw the attention of the reader to some recent highlights in the field.ABPs targeting hydrolases
Hydrolases that act via the formation of a covalent enzyme–substrate intermediate are prime targets for covalent labelling using ABPs. Amongst the hydrolases, proteases continue to be a major focus for novel ABPs, likely due to the plethora of broad-spectrum warheads available against this enzyme class.21 Notably, recent work from the Overkleeft group used epoxyketone ABPs to demonstrate that the proteasome, a multi-protein complex responsible for most regulated protein degradation in the cell, recognises glycosylated substrates.40 This is an observation that may have widespread consequences given the importance of protein glycosylation in immunity, signalling and cellular recognition. The same group has recently elaborated ABPs against cathepsins with a mannose moiety, and shown that they can target professional antigen presenting cells that display cell surface mannose receptors.19 Combining the power of ABPs with such systems for targeted drug delivery has enormous potential significance for the application of ABPs in complex physiological systems,41 and the coming years are likely to see further ground-breaking examples. Several recent reports have extended the range of important hydrolases that can be analysed using ABPs, including dimethylarginine dimethylaminohydrolases,42 phosphatases43–45 postproline proteases,46 histone deacetylases,47 and specific subunits of the proteasome.48 Broad-spectrum ABPs also continue to provide striking insights into basic biology and pathogenesis, for example in the identification of tumour-associated serine hydrolases.6ABPs for hydrolases involved in the virulence of pathogenic microbes also continue to be the subject of intensive study. In addition to the example from C. difficile described above,13 recent reports from the Sieber and Vocadlo labs describe the first such probes for a range of lactamases in MRSA49 and glucosaminidases in Vibrio cholera,50 respectively. The Bogyo and Boothroyd labs have recently reported a remarkably simple ABP based on an irreversible phospholipase inhibitor, 4-bromo phenacyl bromide, which in combination with knockout strains has been used to highlight a large number of enzymes involved in invasion by the apicomplexan pathogen Toxoplasma gondii.51 The interested reader is referred to an excellent recent review by Puri and Bogyo covering ABP developments in the microbial pathogenesis field.52
ABPs targeting other enzyme classes
ABPs that exploit the mechanism of enzymes other than hydrolases are less numerous, though notable advances have been made in recent years. For example, a recent report from the Cravatt lab describes a panel of ABPs capable of labelling multiple cytochrome P450s with a range of specificity, including several P450s with poorly-characterised functions.53 This is a another good example of ABPs providing de novo data about enzyme activity within living systems, thus taking into account a plethora of effects that are not observed in cell-free systems. In collaboration with the Burkart lab, the same group has also reported a set of ABPs targeting polyketide and nonribosomal peptide synthases in bacterial systems, a particularly diverse family of enzymes that are critically important in engineering biosynthetic pathways.54 Ploegh et al. have recently expanded the range of ABPs capable of labelling ubiquitin (Ub) ligases and deubiquitinating enzymes by the simple modification of the C-terminus of Ub with a range of electrophilic traps.55,56 The Ub–proteasome system is the key mediator of targeted protein degradation in the cell, and ubiquitination has also been shown to play an important role in gene regulation and the immune response.57,58 These Ub ABPs complement the wide range of known proteasome ABPs, and have started to provide a much more complete picture of these essential processes. The groups of Yen and Lin have also described the generation of ABPs targeting the biosynthetic activity of cell surface receptor CD38, a key mediator in the immune response,59 and Licht et al. have made the first steps towards ABPs directed at ion channels.60Biotechnological applications of ABPs
In addition to the prominent role ABPs can play in diagnostics and imaging, they provide a powerful mechanism for screening drugs for specificity against multiple targets in vivo. The Cravatt lab has recently extended this paradigm to create a generic technique for screening a specific enzyme using an ABP labelled with a fluorescent dye.61 Upon reaction with the protein, changes in the fluorescence polarisation of the dye due to reduced probe mobility can be used to detect reversible and irreversible inhibitors that compete at the same binding site. This remarkably simple concept is essentially a high-throughput and cell-free variant of the in vivo or live cell competition assays described above. With the explosive growth in the range and variety of ABPs described above, the method has potentially very wide applicability as it does not rely on knowledge regarding a function or a substrate for the enzyme. All that is required is identification of an ABP for the general catalytic class along with access to the purified protein, although with more specific ABPs it may also prove possible to use crude enzyme preparations.A very unusual example of a ‘structure-based’ probe was recently disclosed by Kelly et al.62 In this instance, a stilbene analogue was reported to modify a single lysine side chain (amongst eight) of a single protein, transthyretin, against a background of >4000 other proteins present in blood plasma. Although this labelling does not appear to rely on enzymatic activity, exploiting the unique chemistry of a single residue in a protein has strong parallels to ABP and AfBP technologies.
Conclusions
As the focus of research in organic chemistry continues to shift towards the interface with other disciplines, a platform of chemistry-based technologies is emerging with widespread implications for the life sciences and medicine. Under the general banner of chemical biology, chemical genetics63,64 and chemical proteomics65 have assumed particular prominence, coupled with general approaches for the chemical labelling of biomolecules in vivo.66 ABPP draws on aspects of each of these fields, combining the power of chemical inhibitors with the versatility of bioorthogonal ligation chemistries, and the ever-increasing resolution of modern proteomic techniques.The coming years will see the extension of ABP technology to a growing range of enzyme classes, and the multiplexing of multiple probe types to analyse several activities in one experiment. In particular, due to their importance in signalling and gene regulation, we are likely to see many more ABPs directed against kinases67 and transferases, as well as against their corresponding hydrolases (e.g. phosphatases44 glycosidases,68 and thioesterases69). For example, the elegant bioorthogonal tools recently developed for the identification of transferase substrates would be well-complemented by ABPs directed against the corresponding enzymes.70,71 A range of powerful recombinant and small molecule probes exist for the specific and general detection of enzyme activity (without enzyme labelling),72 and their use in concert with ABPs may prove to be a powerful combination. There are also opportunities for the tighter coupling of ABPs with substrate identification, and recent reports have described the first steps towards this goal.73 Addressing the challenge of enzyme activity associated with sub-cellular localisation or specific complex formation is a frontier in catalomics that will demand greater refinement of current technology, probably in step with the development of novel allosteric inhibitors and in vivo labelling strategies.
The future of catalomics will depend in large part on the capacity of bioorganic chemists to innovate in probe design and the optimisation and application of probes in living systems. Whether this is achieved by interdisciplinary collaboration or by multidisciplinary labs, organic chemistry is set to make a decisive contribution to the exploration of biocatalysis, and to our ability to comprehend and manipulate living systems.
References
- B. F. Cravatt, G. M. Simon and J. R. Yates, 3rd, Nature, 2007, 450, 991–1000 CrossRef CAS.
- K. A. Kalesh, H. Shi, J. Ge and S. Q. Yao, Org. Biomol. Chem., 2010, 8, 1749–1762 RSC.
- A. C. Rios and Y. Tor, Curr. Opin. Chem. Biol., 2009, 13, 660–668 CrossRef CAS.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS.
- A. Shen, P. J. Lupardus, V. E. Albrow, A. Guzzetta, J. C. Powers, K. C. Garcia and M. Bogyo, Nat. Chem. Biol., 2009, 5, 469–478 CrossRef CAS.
- M. Bogyo, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2379–2380 CrossRef CAS.
- M. Uttamchandani, C. H. Lu and S. Q. Yao, Acc. Chem. Res., 2009, 42, 1183–1192 CrossRef CAS.
- W. P. Heal and E. W. Tate, Org. Biomol. Chem., 2010, 8, 731–738 RSC.
- E. W. Tate, J. Chem. Biol., 2008, 1, 17–26 Search PubMed.
- H. Shi, K. Liu, A. Xu and S. Q. Yao, Chem. Commun., 2009, 5030–5032 RSC.
- J. A. Blair, D. Rauh, C. Kung, C. H. Yun, Q. W. Fan, H. Rode, C. Zhang, M. J. Eck, W. A. Weiss and K. M. Shokat, Nat. Chem. Biol., 2007, 3, 229–238 CrossRef CAS.
- M. J. Evans and B. F. Cravatt, Chem. Rev., 2006, 106, 3279–3301 CrossRef CAS.
- T. H. Dang, L. Riva Lde, R. P. Fagan, E. M. Storck, W. P. Heal, C. Janoir, N. F. Fairweather and E. W. Tate, ACS Chem. Biol., 2010, 5, 279–285 CrossRef CAS.
- A. Watzke, G. Kosec, M. Kindermann, V. Jeske, H. P. Nestler, V. Turk, B. Turk and K. U. Wendt, Angew. Chem., Int. Ed., 2008, 47, 406–409 CrossRef CAS.
- A. E. Speers and B. F. Cravatt, Chem. Biol., 2004, 11, 535–546 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS.
- L. E. Edgington, A. B. Berger, G. Blum, V. E. Albrow, M. G. Paulick, N. Lineberry and M. Bogyo, Nat. Med., 2009, 15, 967–973 CrossRef CAS.
- U. Hillaert, M. Verdoes, B. I. Florea, A. Saragliadis, K. L. Habets, J. Kuiper, S. Van Calenbergh, F. Ossendorp, G. A. van der Marel, C. Driessen and H. S. Overkleeft, Angew. Chem., Int. Ed., 2009, 48, 1629–1632 CrossRef CAS.
- A. E. Speers and B. F. Cravatt, J. Am. Chem. Soc., 2005, 127, 10018–10019 CrossRef CAS.
- W. P. Heal, S. R. Wickramasinghe and E. W. Tate, Curr. Drug Discovery Technol., 2008, 5, 200–212 Search PubMed.
- D. K. Nomura, J. Z. Long, S. Niessen, H. S. Hoover, S. W. Ng and B. F. Cravatt, Cell (Cambridge, Mass.), 2010, 140, 49–61 CrossRef CAS.
- J. A. Menendez and R. Lupu, Nat. Rev. Cancer, 2007, 7, 763–777 CrossRef CAS.
- Y. Liu, M. P. Patricelli and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14694–14699 CrossRef CAS.
- M. P. Washburn, D. Wolters and J. R. Yates, 3rd, Nat. Biotechnol., 2001, 19, 242–247 CrossRef CAS.
- K. P. Chiang, S. Niessen, A. Saghatelian and B. F. Cravatt, Chem. Biol., 2006, 13, 1041–1050 CrossRef CAS.
- J. Z. Long, W. Li, L. Booker, J. J. Burston, S. G. Kinsey, J. E. Schlosburg, F. J. Pavon, A. M. Serrano, D. E. Selley, L. H. Parsons, A. H. Lichtman and B. F. Cravatt, Nat. Chem. Biol., 2009, 5, 37–44 CrossRef CAS.
- J. C. Reed, Cancer J. Sci. Am., 1998, 4(Suppl 1), 8–14 Search PubMed.
- S. Launay, O. Hermine, M. Fontenay, G. Kroemer, E. Solary and C. Garrido, Oncogene, 2005, 24, 5137–5148 CrossRef CAS.
- V. Ntziachristos, E. A. Schellenberger, J. Ripoll, D. Yessayan, E. Graves, A. Bogdanov, Jr., L. Josephson and R. Weissleder, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12294–12299 CrossRef CAS.
- D. Kato, K. M. Boatright, A. B. Berger, T. Nazif, G. Blum, C. Ryan, K. A. Chehade, G. S. Salvesen and M. Bogyo, Nat. Chem. Biol., 2005, 1, 33–38 CrossRef CAS.
- M. Green and P. M. Loewenstein, Cell (Cambridge, Mass.), 1988, 55, 1179–1188 CrossRef CAS.
- K. Zavitsanou, V. Nguyen, I. Greguric, J. Chapman, P. Ballantyne and A. Katsifis, J. Mol. Histol., 2007, 38, 313–319 Search PubMed.
- C. Adams, K. Totpal, D. Lawrence, S. Marsters, R. Pitti, S. Yee, S. Ross, L. Deforge, H. Koeppen, M. Sagolla, D. Compaan, H. Lowman, S. Hymowitz and A. Ashkenazi, Cell Death Differ., 2008, 15, 751–761 CrossRef CAS.
- G. Blum, R. M. Weimer, L. E. Edgington, W. Adams and M. Bogyo, PLoS One, 2009, 4, e6374 CrossRef.
- E. J. Kuipers and C. M. Surawicz, Lancet, 2008, 371, 1486–1488 CrossRef.
- T. Schirmeister and A. Klockow, Mini-Rev. Med. Chem., 2003, 3, 585–596 CrossRef CAS.
- A. F. Berry, W. P. Heal, A. K. Tarafder, T. Tolmachova, R. A. Baron, M. C. Seabra and E. W. Tate, ChemBioChem, 2010, 11, 771–773 CrossRef CAS.
- J. M. Kirby, H. Ahern, A. K. Roberts, V. Kumar, Z. Freeman, K. R. Acharya and C. C. Shone, J. Biol. Chem., 2009, 284, 34666–34673 CrossRef CAS.
- M. D. Witte, B. I. Florea, M. Verdoes, O. Adeyanju, G. A. Van der Marel and H. S. Overkleeft, J. Am. Chem. Soc., 2009, 131, 12064–12065 CrossRef CAS.
- E. A. Raiber, C. Tulone, Y. Zhang, L. Martinez-Pomares, E. Steed, A. M. Sponaas, J. Langhorne, M. Noursadeghi, B. M. Chain and A. B. Tabor, ACS Chem. Biol., 2010, 5, 461–476 CrossRef CAS.
- Y. Wang, S. Hu and W. Fast, J. Am. Chem. Soc., 2009, 131, 15096–15097 CrossRef CAS.
- C. C. Kuo, C. Y. Chu, J. J. Lin and L. C. Lo, Biochem. Biophys. Res. Commun., 2010, 391, 230–234 CrossRef CAS.
- K. A. Kalesh, L. P. Tan, K. Lu, L. Gao, J. Wang and S. Q. Yao, Chem. Commun., 2010, 46, 589–591 RSC.
- K. Shen, L. Qi, M. Ravula and K. Klimaszewski, Bioorg. Med. Chem. Lett., 2009, 19, 3264–3267 CrossRef CAS.
- E. Sabido, T. Tarrago, S. Niessen, B. F. Cravatt and E. Giralt, ChemBioChem, 2009, 10, 2361–2366 CrossRef CAS.
- C. M. Salisbury and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1171–1176 CrossRef CAS.
- C. Gu, I. Kolodziejek, J. Misas-Villamil, T. Shindo, T. Colby, M. Verdoes, K. H. Richau, J. Schmidt, H. S. Overkleeft and R. A. van der Hoorn, Plant J., 2009, 62, 160–170.
- I. Staub and S. A. Sieber, J. Am. Chem. Soc., 2009, 131, 6271–6276 CrossRef CAS.
- K. A. Stubbs, A. Scaffidi, A. W. Debowski, B. L. Mark, R. V. Stick and D. J. Vocadlo, J. Am. Chem. Soc., 2008, 130, 327–335 CrossRef CAS.
- S. Ravindran, M. B. Lodoen, S. H. Verhelst, M. Bogyo and J. C. Boothroyd, PLoS One, 2009, 4, e8143 CrossRef.
- A. W. Puri and M. Bogyo, ACS Chem. Biol., 2009, 4, 603–616 CrossRef CAS.
- A. T. Wright, J. D. Song and B. F. Cravatt, J. Am. Chem. Soc., 2009, 131, 10692–10700 CrossRef CAS.
- J. L. Meier, S. Niessen, H. S. Hoover, T. L. Foley, B. F. Cravatt and M. D. Burkart, ACS Chem. Biol., 2009, 4, 948–957 CrossRef CAS.
- K. R. Love, R. K. Pandya, E. Spooner and H. L. Ploegh, ACS Chem. Biol., 2009, 4, 275–287 CrossRef.
- H. Ovaa, Nat. Rev. Cancer, 2007, 7, 613–620 CrossRef CAS.
- Y. C. Liu, Annu. Rev. Immunol., 2004, 22, 81–127 CrossRef CAS.
- A. Shilatifard, Annu. Rev. Biochem., 2006, 75, 243–269 CrossRef CAS.
- H. Jiang, J. Congleton, Q. Liu, P. Merchant, F. Malavasi, H. C. Lee, Q. Hao, A. Yen and H. Lin, J. Am. Chem. Soc., 2009, 131, 1658–1659 CrossRef CAS.
- M. Tantama, W. C. Lin and S. Licht, J. Am. Chem. Soc., 2008, 130, 15766–15767 CrossRef CAS.
- G. M. Simon and B. F. Cravatt, J. Biol. Chem., 2010, 285, 11051–11055 CrossRef CAS.
- S. Choi, S. Connelly, N. Reixach, I. A. Wilson and J. W. Kelly, Nat. Chem. Biol., 2010, 6, 133–139 CAS.
- K. Shah, Y. Liu, C. Deirmengian and K. M. Shokat, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 3565–3570 CrossRef CAS.
- B. R. Stockwell, Nat. Rev. Genet., 2000, 1, 116–125 CrossRef CAS.
- D. A. Jeffery and M. Bogyo, Curr. Opin. Biotechnol., 2003, 14, 87–95 CrossRef CAS.
- S. Chattopadhaya, F. B. Abu Bakar and S. Q. Yao, Curr. Med. Chem., 2009, 16, 4527–4543 CrossRef CAS.
- K. A. Kalesh, D. S. Sim, J. Wang, K. Liu, Q. Lin and S. Q. Yao, Chem. Commun., 2010, 46, 1118–1120 RSC.
- C. S. Tsai, Y. K. Li and L. C. Lo, Org. Lett., 2002, 4, 3607–3610 CrossRef CAS.
- S. J. Kridel, F. Axelrod, N. Rozenkrantz and J. W. Smith, Cancer Res., 2004, 64, 2070–2075 CrossRef CAS.
- W. P. Heal, S. R. Wickramasinghe, P. W. Bowyer, A. A. Holder, D. F. Smith, R. J. Leatherbarrow and E. W. Tate, Chem. Commun., 2008, 480–482 RSC.
- W. P. Heal, S. R. Wickramasinghe, R. J. Leatherbarrow and E. W. Tate, Org. Biomol. Chem., 2008, 6, 2308–2315 RSC.
- J. D. Violin, J. Zhang, R. Y. Tsien and A. C. Newton, J. Cell Biol., 2003, 161, 899–909 CrossRef CAS.
- S. Suwal and M. K. Pflum, Angew. Chem., Int. Ed., 2010, 49, 1627–1630 CAS.
| This journal is © The Royal Society of Chemistry 2011 |