Food analysis and food authentication by peptide nucleic acid (PNA)-based technologies
Stefano Sforza
*,
Roberto Corradini
,
Tullia Tedeschi
and
Rosangela Marchelli
Department of Organic and Industrial Chemistry, University of Parma, Parco Area delle Scienze 17a, University Campus, I-43124, Parma, Italy. E-mail: stefano.sforza@unipr.it; Fax: +39 0521-905472; Tel: +39 0521-905406
First published on 30th September 2010
Abstract
This tutorial review will address the issue of DNA determination in food by using Peptide Nucleic Acid (PNA) probes with different technological platforms, with a particular emphasis on the applications devoted to food authentication. After an introduction aimed at describing PNAs structure, binding properties and their use as genetic probes, the review will then focus specifically on the use of PNAs in the field of food analysis. In particular, the following issues will be considered: detection of genetically modified organisms (GMOs), of hidden allergens, of microbial pathogens and determination of ingredient authenticity. Finally, the future perspectives for the use of PNAs in food analysis will be briefly discussed according to the most recent developments.
 Stefano Sforza | Stefano Sforza is an Associate Professor of Organic and Food Chemistry at the Faculty of Agriculture of the University of Parma. His main research fields involve the synthesis and study of new chiral modified Peptide Nucleic Acids (PNAs) and their binding properties to DNA and RNA, the use of PNAs for food analysis, the development of new mass spectrometry-based methods for the structural determination and analysis of biomolecules (amino acids, peptides, toxins, etc.) in foods, the assessment of food authenticity through the use of DNA and peptides as molecular markers. |
 Roberto Corradini | Roberto Corradini is an Associate Professor in Organic and Bioorganic Chemistry in the Biotechnology Degree at the University of Parma. He is a member of the Editorial board of Chirality (Wiley, VCH). His research work has been mainly in the field of molecular recognition and enantiomer discrimination of biological molecules. In particular, he has contributed to the development of cyclodextrin based sensors and peptide nucleic acid (PNA) monomers and oligomers and of PNA-based methods for identification of DNA sequences, both in biomedicine and in food analysis. |
 Tullia Tedeschi | Tullia Tedeschi is a Researcher in Organic Chemistry at the Faculty of Agriculture of the University of Parma. Her research interests are focused on the synthesis of chiral Peptide Nucleic Acids (PNA) with high optical purity, study of their interaction with complementary DNA and RNA by spectrophotometrical techniques and development of new methodologies (microarrays, capillary electrophoresis and gel-electrophoresis) for using PNAs as highly specific genetic probes. |
 Rosangela Marchelli | Rosangela Marchelli is a full professor of Organic Chemistry at the Faculty of Agriculture of the University of Parma, and delegate of the Italian Chemical Society in the Division of Food Chemistry of EuCheMS (European Chemical and Molecular Sciences). She is also a member of the NDA (Nutrition, Dietetics and Food Allergy) Panel of EFSA (European Food Safety Authority). Her research activity focuses on DNA and RNA recognition by means of chiral peptide nucleic acids (PNAs), determination of peptides in food by HPLC/MS and the development of new detection methods of mycotoxins in food. |
1. Introduction
The research on new and selective methods and technologies for fast, reliable and sensitive detection of specific DNA sequences has important applications in many fields. In food analysis, DNA detection is increasingly applied as an answer to different needs, such as for GMO detection,1 microbial pathogen determination,2 assessment of the presence of undeclared allergenic ingredients.3The detection of DNA sequences in any matrix always starts with its extraction and several kits are available on the market. Extraction procedures have to be carefully optimized, since this step can be very difficult in food, due to the complexity of the matrices and in some cases, such as oils or lecithin, to the low abundance of DNA.4 Amplification of the target is normally performed by Polymerase Chain Reaction (PCR) using specific primers in order to amplify selectively the region of interest of the DNA. The presence of an amplified DNA product of the expected size, normally assessed by gel electrophoresis, is usually considered a sufficient proof of the presence of the sequence of interest. Real time PCR techniques are used for DNA quantification, and also in this case the appearance of a fluorescent signal, linked to the presence of an amplified DNA sequence, is usually considered sufficient for proving the presence of the DNA sequence of interest. In these cases, the selectivity and the specificity of DNA detection heavily relies on the selectivity and the specificity of the primers used for the DNA amplification.5
Nevertheless, in order to avoid false positives or when similar DNA sequences have to be discriminated, in many cases the confirmation of the identity of the amplified DNA is considered necessary.6 This can be achieved by “nested PCR”, which consists in a further amplification of a shorter sequence within the former strand.5 Alternatively, recognition of the presence of the target sequence can be performed by hybridization with specific probesvia Watson–Crick base pairing. Recognition should be as specific as possible, and must be followed by a change in some measurable property either in solution or on a sensing surface. This can be achieved by subsequent reactions (mostly enzymatic) or by changes in some physico-chemical property.
In the field of the molecular probes, Peptide Nucleic Acids (PNAs) can be considered very promising tools due to their unique properties. PNAs are DNA mimics in which the negatively charged sugar–phosphate backbone is replaced by a neutral polyamide backbone composed of N-(2-aminoethyl) glycine units (Fig. 1).7 In the PNA structure, in spite of the different chemical functionalities, the backbone length and the distance of the nucleobases from the backbone are the same as in natural DNA. As a consequence, PNAs can bind complementary DNA or RNA sequences following standard Watson–Crick rules.8 Due to the lack of electrostatic repulsions, given the uncharged nature of the PNA backbone, PNA/DNA and PNA/RNA complexes have improved thermal stability as compared to DNA/DNA and DNA/RNA duplexes. The high affinity for the DNA target allows to decrease the limits of detection in many applications devoted to DNA recognition.
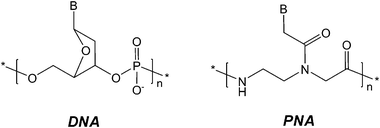 | ||
| Fig. 1 Chemical structures of DNA and PNA. | ||
Another interesting feature of PNA probes is their selectivity: a single base-pairing mismatch in PNA/DNA duplexes is more destabilizing than in DNA/DNA complexes of the same length. Moreover, since the increased affinity allows us to obtain stable PNA–DNA and PNA–RNA complexes even with short PNA probes (10-mer or less), as compared to DNA probes, the possibility to use short probes in post-PCR applications favours an even improved selectivity, which in some applications allows for an easy detection of single-point mutations and single nucleotide polymorphisms.9
A further outstanding advantage of PNA probes arises from the fact that, because of the neutrality of the PNA backbones, PNA–DNA hybridization is less sensitive to the presence of salts, which are necessary to attenuate electrostatic repulsions in duplex DNA. Actually, PNA–DNA binding can be efficiently achieved even under very low salt concentrations, a condition that promotes the destabilization of RNA and DNA secondary structures, resulting in an improved access to target sequences.10 Low ionic strength conditions are essential when targeting a double-strand DNA, in order to disfavour DNA–DNA duplex formation, and to allow PNA probes to invade the double helix, displacing the homologous DNA strand. The relative independence of their performance from the environment makes the analytical procedures more robust, especially in the case of the analysis of complex matrices such as foods.
Finally, another interesting property of PNAs, which is useful in many biological applications, is their stability to both nucleases and peptidases, since their “unnatural” skeleton prevents recognition by natural enzymes, making them highly persistent in biological fluids.11 A major drawback is that enzymatic reactions, which are often used in combination with DNA probes, are not possible using PNA substitutes. Therefore, detection schemes involving e.g. DNA-ligases or DNA-polymerases cannot be performed with PNAs. However, PNAs can easily be modified with recognition elements for proteins (such as biotin) and then coupled with enzymatic assays. As a further consequence of their enzymatic and chemical stability, kits and sensory systems based on PNA probes are superior to DNA-based ones for long-term storage.
The excellent hybridization properties of PNA oligomers, combined with their unique chemistry, have been exploited in a variety of molecular biology tools, biomedical applications and diagnostic techniques.12 The scarce commercial availability of PNA probes, in the last years a limiting step, is being overcome by the fact that custom-made PNA probes are now commercially available, being easily synthesized by the standard procedures usually adopted in peptide chemistry. The high chemical stability of PNAs allows the use of several different protecting groups for the terminal nitrogen (including Fmoc and Boc) and for the nucleobases (amides, Cbz or acid labile protecting groups), fully exploiting the different strategies developed in the last years in peptide synthesis.13
In food analysis, although still not exploited routinely, several promising applications have been published in the last years. PNA probes have been used in different applications according to the detection schemes outlined in Fig. 2: (i) as inhibitors of PCR reactions for enhancing the specificity, such as in PCR clamping;14 (ii) as probes able to modify the properties of the target DNA in separation techniques (such as electrophoresis or liquid chromatography) or in optical detection (for example using cyanine dyes which are known to aggregate on PNA:DNA hybrids undergoing a change in colour) (Fig. 2d); (iii) as fluorescently labelled PNA probes in applications where the target nucleic acid can be isolated and subsequently washed in order to remove the excess of the free probe, such as in PNA-FISH,15 or with labelled PNA switching probes (beacons) useful in real-time detection (Real-time PCR)16 (Fig. 2e) and HPLC; (iv) in surface applications, as capture probes for labelled DNA deriving from PCR amplification, as in PNA-microarrays,17 or for unlabelled DNA in the so called “label free” techniques (Fig. 2f).
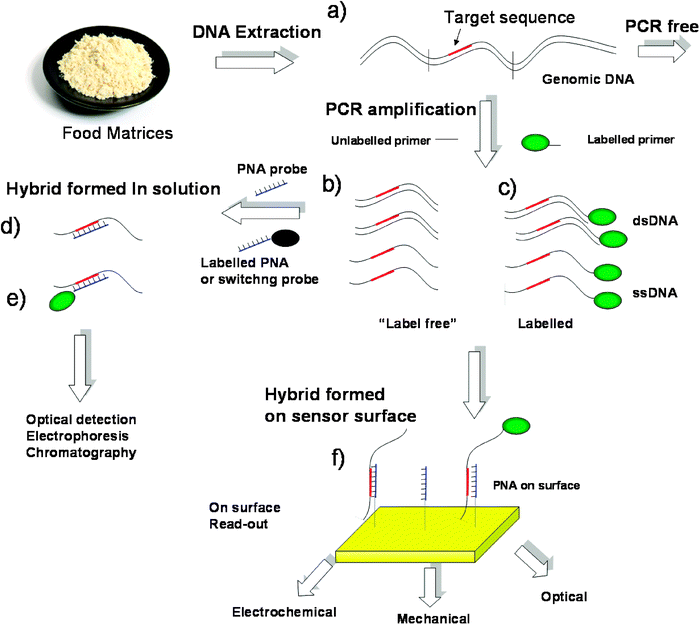 | ||
| Fig. 2 Protocols for DNA analysis by PNA-based technologies. (a) Extraction of DNA from food matrices; (b) a DNA sequence containing the target sequence can be amplified by PCR using specific unlabelled primers; according to the conditions used, only dsDNA or a mixture of ds- and ssDNA can be obtained; (c) amplification of DNA with labelled primers gives labelled PCR products; (d) hybridization in solution with unlabelled PNA; (e) hybridization in solution with PNA switching probes; (f) hybridization on a surface carrying PNA catching probes. | ||
2. GMO determination
Food safety and complete food characterization through traceability are at present one of the major issues in Health and Consumer protection. In particular, in the European Union (EU), labelling of food containing more than 0.9% of GMOs is required and regulations about food and feed labelling and traceability are in force.18 This is motivated by consumers' needs for information and protection, but on the other side it implies more onerous and cost-effective procedures for the food industry. Technical implementation of tools and procedures for product traceability can greatly help to simplify analysis and reduce costs, thus making the labelling procedures more “producers' friendly”.A very important achievement of analytical systems, which can be used for GMO traceability, is the possibility to recognize living organisms through the identification of DNA, a technique which has received in the past decade a tremendous momentum from the scientific efforts linked to the research in genomics. There has been an increasing interest in the research for the application of genetic tools to the traceability of components of interest in the food chain, and standardized procedures are now available for the detection of specific DNA sequences, as well as many laboratories can now provide validated methods for the detection of a wide range of GMOs, based on semiquantitative or quantitative polymerase chain reaction (PCR) procedures.19 Several private laboratories are currently running thousands of DNA analyses per year and this type of market is constantly increasing, not only to comply with the European regulations, but also to provide proper information to consumers. Actually, although PCR methods have been validated for several matrices, the ease of applicability of this type of analyses can further be improved. For the quantitative determination of GMOs the proper standard material is required, which is not always available. The use of highly specific probes for the confirmation of the identity of PCR products is particularly important where the presence of GMOs has immediate industrial or legal consequences (for example, several years ago, hundreds of hectares of maize contaminated with small percentages of GM varieties were destroyed in Northern Italy).20 Thus, the use of highly specific probes either in the post-PCR assessment or as components of biosensors is highly desirable in these cases. On account of their properties, PNAs have been used for the detection of specific sequences in advanced diagnostic methods for the detection of GMOs.
One very simple and immediate method of assessing electrophoretic band identity was the use of the so called “PCR clamping” technique, which was originally proposed for the detection of point mutations.14 By adding a PNA able to bind next to the primer site on the target DNA, the specific inhibition of the PCR product, as revealed by agarose gel analysis, was obtained, allowing qualitative and semi-quantitative determination of the GMO presence.21 This method is particularly suitable when no other equipment except the simple and cheap gel electrophoresis apparatus is available.
In a more complex approach, PNA probes linked to microtitre plates were used to specifically capture biotin-labelled target DNA sequences, which were then used as template in real time immuno-PCR reactions. High sensitivity (LOD in the attomolar range) was reported for MON810 maize using this method.22
Another common equipment present in many laboratories is HPLC. Hybridization of oligonucleotides with PNA can be detected using an anion exchange column, operating under non-denaturing conditions.23 A protocol using PNA hybridization and anion exchange HPLC was developed for the assessment of the identity of PCR products derived from genetically modified soybean and maize. PNAs of different lengths aimed at evaluating the effect of the PNA structure on signal intensity and specificity were used for gene sequences corresponding to a region of the CaMV 35S-CTP construct of Roundup Ready (RR) GM-soybean and to the CryIA sequence of the Bt-176 GM-maize. The results showed that it was possible to perform a clear identification of the PCR product based on the presence of the chromatographic peak of the PNA:DNA hybrid (Fig. 3A).24 In this study, the PCR product was fluorescently labelled and, in order to allow access of the PNA to its target, an excess of one of the two strands had to be generated either by an enzymatic digestion or by asymmetric PCR.
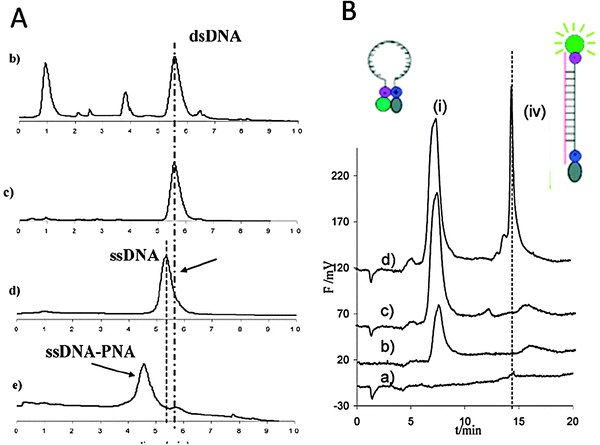 | ||
| Fig. 3 IE HPLC profiles obtained using: (A) an unlabelled PNA probe and labeled DNA; (B) a PNA beacon and unlabelled DNA. (A) HPLC profile of a Cy5-labelled PCR amplicon specific for RR soybean (79 bp) obtained from a soy burger labelled “GMO-free”: (b) the PCR product (dsDNA) crude; (c) dsDNA after purification, (d) after digestion with λ-exonuclease (ssDNA); (e) after hybridization with a specific PNA probe (ssDNA–PNA). Column: TSK gel DEAE–NPR (4.6 mm id × 3.5 cm). T = 35 °C. Eluent A: Tris 20 mM in H2O at pH 9; eluent B: NaCl 1 M in eluent A at pH 9. Gradient: from 50% A to 30% A in 10 min. Flow rate: 0.5 mL min−1. Fluorescence detector: λex = 646 nm, λem = 664 nm. (reproduced with permission from ref. 24). (B) HPLC profile of the same PCR product as in (A) unlabelled (from soy flour containing RR-soy). (a) PCR product alone; (b) PNA beacon (1 μM) alone; (c) PNA beacon (1 μM) + a non-specific PCR product; (d) PNA beacon (1 μM) + the specific PCR product. Attribution: (i) beacon and components of the PCR reaction; (iv) PNA beacon–DNA hybrid. T = 25 °C. Column: TSK-gel DNA NPR (4.6 mm id × 7.5 cm); eluent A: Tris 0.02 M, pH = 9.0, eluent B: NaCl 1 M in eluent A. Linear gradient: from 100% A to 100% B in 20 min; flow rate: 0.5 mL min−1. λex = 497 nm, λem = 520 nm (reproduced with permission from ref. 26). | ||
PNA labelled with fluorophores can be alternatively used to trace DNA with no need of labelling the target DNA. However, the background fluorescence of the free probe usually prevents the specific detection and quantitation of the PNA:DNA hybrid. In an impressive demonstration of the PNA properties, a single-molecule detection of transgenic DNA was performed in early studies by means of PNA probes and double wavelength fluorescence analysis.25 In this study two PNA probes complementary to two different sequences of the transgene, labelled with different fluorophores, were used in connection with an advanced apparatus composed of two single-photon counting detectors. The simultaneous detection of the PNA probes combined with the target DNA was performed, thus allowing to discard signals due to the free probes. Subsequent studies however were devoted to develop methods which could be used with more common equipment available in analytical laboratories.
PNA beacons and the related “light up probes” have also been produced, in analogy with molecular beacon oligonucleotides. They are modified with a fluorophore and a quencher (or a quenching surface) at each end, held together by hydrophobic interactions in aqueous solution; upon interaction with the target DNA a fluorescence “switch-on” occurs (Fig. 3B). PNA beacons present, relatively to DNA beacons, the advantages of a higher selectivity and a simpler design. One of the major limitations in diagnostics is represented by the eventual fluorescence background of the free (uncomplexed) beacon, which, though lower than that of a fluorescently labelled probe, can, however, interfere with the signal obtained by the PNA:DNA duplex with the analyte sequence at low concentrations, especially for DNA amplified from complex matrices such as food. A way for overcoming this problem was the combined use of PNA beacons and IE-HPLC for the selective label-free detection of DNA, taking advantage of the very specific signal generated by the duplex DNA:PNA beacon, which allows to avoid the presence of unspecific peaks. This approach was used to detect label-free DNA amplified from Roundup Ready soy in a pilot experiment (Fig. 3B).26 A PNA beacon containing a chiral monomer modified at C5 with a lysine side chain was shown to perform a better label-free DNA recognition in a model system.27
Microarray technology is a very powerful tool for the simultaneous detection of several DNA sequences and multi-samples analysis. The use of PNA probes by microarray platforms with fluorescence read-out has been successfully used in the detection of GMOs. Since the detection limit of this technique is in the nanomolar range, multiple PCR amplification, with Cy3 or Cy5 labelling of the target DNA, is performed prior to hybridization. Procedures for obtaining a single stranded DNA have to be used in this case, either by enzymatic digestion of one strand or by asymmetric PCR (i.e. using an excess of one primer).
A PCR protocol allowing for the simultaneous detection of five transgenes and two endogenous controls in food and feed matrices was developed28 and procedures for the deposition of PNAs on microarrays were optimized and proved to be suitable for GMO detection.29 The two methods were combined in order to develop a PNA array device for the screening of GMOs in food. PNA probes complementary to the GMO sequences were opportunely designed, synthesized, and deposited on commercial slides. The best performances were obtained with 15-mer probes and by means of a sufficiently long spacer allowing the PNA probes to be accessible for hybridization with the target DNA. The device was tested on a model system constituted by flour samples containing a mixture of standards at known concentrations of transgenic material, in particular Roundup Ready soybean and Bt11, Bt176, Mon810, and GA21 maize. The DNA was amplified using the specific multiplex PCR method and tested on the PNA array. Every GMO present in the tested samples (Fig. 4) was correctly identified by subsequent fluorescence measurements by a microarray reader.30
 | ||
| Fig. 4 Specificity assessment of the PNA array. Each slide was hybridized with DNA and amplified twice by multiplex PCR, previously extracted from flour containing: (1) GMO free soybean; (2) GMO free maize; (3) 5% MON810 maize; (4) 5% RR soybean; (5) 5% Bt11 maize; (6) 5% Bt176 maize; and (7) 5% GA21 maize. The PNA probes were spotted, at a concentration of 30 μM, as follows: SL (soybean lectin), MZ (maize zein), MON810 (MON810 maize), RR (RR soybean), Bt11 (Bt11 maize), Bt176 (Bt176 maize), and GA21 (GA21 maize) (reproduced with permission from ref. 30). | ||
An advanced technology for the detection of transgenic DNA using fluorescence detection was described by Knoll and coworkers using Surface Plasmon enhanced Fluorescent Spectroscopy (SPFS), which enables us to measure selectively the fluorescence of molecules captured on the sensor surface by excitation through an evanescent field generated in the SPR experiment.31 High sensitivity and excellent mismatch recognition was demonstrated in these studies.32
Sandwich hybridization assays with gold nanoparticles and Surface Plasmon Resonance Imaging (SPRI) readout were used for improving detection sensitivity for oligonucleotide hybridization down to low femtomolar concentrations. This method uses a nanoparticle-enhanced SPRI detection scheme based on PNA probes (Fig. 5).33 The method was successfully used for the discrimination between oligonucleotides matching the PNA probe sequence and oligonucleotides carrying a single-base mismatch. The same method was subsequently used for the direct detection of GMO in a “PCR-free” analysis, using genomic DNA extracted from soy flour. Avoiding amplification by PCR would be a great advantage, since in this way less steps are required in the overall procedure with less occasions for contaminations. This methodology gave striking performances, since quantities as low as 0.1% of GMO material in samples containing 10 pg μL−1 gave significant signals in the presence of a large excess of non-target DNA.34 Targeting of large dsDNA was facilitated by sonication, which broke the DNA into smaller fragments, and by a hybridization protocol which used denaturation at high temperature, followed by freezing of the sample in order to avoid reannealing of DNA. The use of microfluidic channels allowed then to capture the target sequence and wash out the non-complementary strand. The high specificity was due to a combination of different events: (a) capture of DNA by the highly specific PNA probes; (b) efficient capture of the oligonucleotide-labelled gold nanoparticles which catalyzed the subsequent aggregation of other nanoparticles, thus amplifying the optical signal.
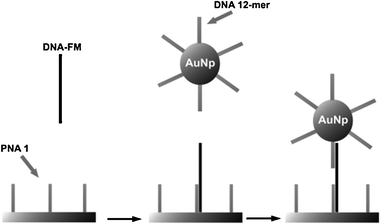 | ||
| Fig. 5 Description of the strategy used for the ultrasensitive nanoparticle-enhanced SPRI detection of the target DNA sequence. PNA 1: surface immobilized specific PNA capture probe; DNA-FM: DNA full match to be detected; DNA 12-mer: specific DNA capture probe linked to gold nanoparticles; AuNp: gold nanoparticles (reproduced with permission from ref. 33). | ||
The possibility to avoid PCR and to detect directly the genomic DNA can be very useful in food as well as in biomedical applications when the target DNA is present at very low concentrations.
3. Hidden allergen determination
Food allergy is an expanding concern in western countries, as severe reactions to foods have become more common in the recent years.35 Recently, in order to protect the consumers, the EU introduced a directive which lists 14 common allergenic ingredients to be declared on the label when present in a given food (Directive 2007/68/EC). Nevertheless, of particular concern are the so called “hidden allergens”, i.e. allergenic ingredients accidentally present in a food, and thus not declared on the label, which may trigger severe allergic reactions if inadvertently consumed by susceptible subjects. Although direct detection methods of allergenic proteins are usually the first choice for screening purposes, indirect methods, based on the detection of DNA sequences specific for a given allergenic ingredient, are also becoming popular.36 Actually, direct methods can fail when applied to complex food mixtures or to severely processed foods, in which proteins may be heavily modified and, therefore, not detectable by antibodies directed to recognize their native forms. In contrast, DNA detection is usually more feasible in these cases, DNA being more resistant to drastic thermal treatments. However, the low amount of DNA present implies that a very specific confirmatory analysis is usually mandatory. Peptide Nucleic Acids, thanks to their very specific DNA binding properties, their chemical and enzymatic stability and the possibility to be used in connection with several detection methods, are ideal candidates for the task.The possibility to use PNAs as confirmatory probes post-PCR analysis has been first demonstrated in combination with HPLC.37 A PCR analysis was developed aimed at amplifying a 156 bp region of the gene coding for Cor a 1, the major hazelnut allergen. Simultaneously, a 15-mer PNA probe complementary to an internal region of the amplicon was designed and synthesized, and used in anion exchange HPLC. When targeting a double stranded DNA produced in a PCR reaction, the major problem hampering the detection by PNA probes is represented by the necessity to invade the DNA double helix. In fact, since DNA recognition by PNAs takes place via standard Watson–Crick hydrogen bonds, the PNA probe must be able to compete with the homologous DNA strand for binding to the target region. In order to circumvent this problem, the single strand PCR product was obtained, by using a suitable PCR primer, labelled at the 5′ terminus in the strand homologous to PNA (the one not to be targeted by the PNA probe) with a phosphate group. The post-PCR selective enzymatic digestion of the strand functionalized with the phosphate with Lambda exonuclease allowed PNA to bind to the remaining single strand DNA. The second major problem, i.e. the sensitivity of the detection system for the PNA–DNA binding event, was solved by labelling, again with the use of a suitable PCR primer, the remaining strand with a fluorophore, such as the Cy5 dye. The definitive confirmatory experiment was then obtained by a set of four injections in the HPLC-FLD system, as represented in Fig. 6. First, the double strand PCR product was injected and detected (Fig. 6a). Then, the PCR product after enzymatic digestion was analysed: as it can be seen, the single strand DNA showed a chromatographic shift as compared to the double strand DNA (still present in small amounts) (Fig. 6b). In the third experiment, the single strand DNA was injected after hybridization with the complementary PNA: the formation of a PNA–DNA duplex could be visualized by a further shift, much more marked, of the retention times (Fig. 6c). Finally, as a final confirmation, the single strand DNA was injected after mixing with a PNA non-complementary to any region of the single strand amplified DNA: in this case the DNA did not change its retention time as compared to the free DNA in the second experiment, clearly showing that no hybridization had taken place (Fig. 6d).37 It is to be remarked that, by using fluorescence detection, the free PNA is not detectable with this system, and its presence can be evidenced only by the effect exerted on the chromatographic behavior of DNA. In this way, excess PNA can be used in the test without any interference with the analysis. By using this methodology, the accidental presence of hazelnut DNA in food matrices not containing hazelnut as ingredient was detected.37
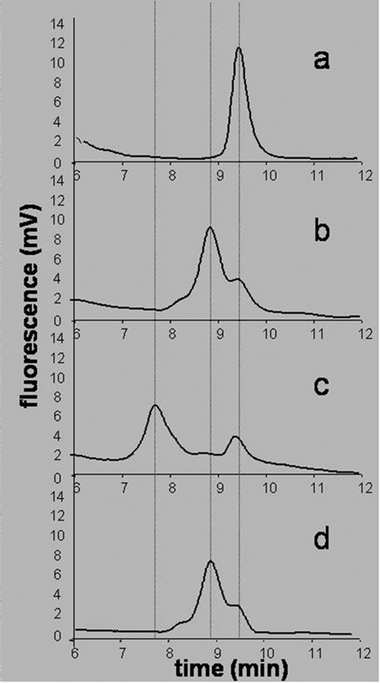 | ||
| Fig. 6 AE-HPLC identity assessment of the PCR amplified DNA of hazelnut (Cor a 1) by a PNA probe: (a) double stranded DNA from PCR; (b) single strand DNA after enzymatic digestion; (c) hybridization of the single strand DNA after enzymatic digestion with the specific PNA probe complementary to the hazelnut DNA sequence; (d) hybridization of the single strand DNA after enzymatic digestion with a PNA probe non-complementary to any hazelnut DNA sequence (reproduced with permission from ref. 37). | ||
The possibility of detecting hidden allergens by PNA-based post-PCR confirmatory analysis was subsequently tackled with a different technique, i.e. by using PNA microarrays, demonstrating that multiple allergen detection is feasible by this platform.38 In this case PNA array platforms were designed by synthesizing two different probes complementary to the gene regions coding for Cor a 1 (the major hazelnut allergen) and Ara h 2 (the major peanut allergen). These PNA probes were synthesized with two 2-(2-aminoethoxy)ethoxyacetyl spacers at the amino terminus, in order to have an unhindered free amino group to be used for linking to the array surfaces. Coupling conditions to the surface were carefully optimized according to the characteristics of PNA chemistry. In particular, given the low solubility of PNAs in aqueous solvents, several washing cycles with different solvents were introduced in order to avoid PNA dragging during deposition. After slide deposition, the sensitivity of the PNA microarrays was tested with synthetic oligonucleotides, allowing to define a limit of detection down to 1 nM DNA (in the reported experimental conditions). Then, a duplex PCR (i.e. a PCR simultaneously amplifying both the genomic region of hazelnuts and peanuts) was developed. The arrays were first tested with amplicons from pure hazelnut and peanut, demonstrating the specificity of the system (Fig. 7). Finally, food products commercially available, purchased on the market, were screened. PCR amplicons obtained from the samples were tested with the PNA microarrays, confirming in some samples the presence of hazelnuts and/or peanuts, in some cases even undeclared either as ingredients or possible contaminants (Fig. 8).38 It must be said that these experiments were performed before the implementation of the actual EU regulation. Thus, PNA microarrays were shown to be suitable platforms for the fast and reliable post-PCR confirmation of the DNA identity in food analysis.
 | ||
| Fig. 7 PNA microarrays tested with PCR products deriving from amplification of DNA extracted from pure hazelnut (a) and peanut (b). H: PNA complementary to hazelnut DNA; P: PNA complementary to peanut DNA; B: blank; CP: control probe (reproduced with permission from ref. 38). | ||
 | ||
| Fig. 8 PNA array analysis for the detection of hazelnut and peanut in commercial foodstuffs. (a): Breakfast cereals, possible traces of tree nuts and peanuts declared; (b): Muesli snack with chocolate, peanut declared, hazelnut NOT declared. H: PNA complementary to hazelnut DNA; P: PNA complementary to peanut DNA; B: blank; CP: control probe (reproduced with permission from ref. 38). | ||
A quite unconventional method using PNA probes for post-PCR confirmation of DNA identity based on circular dichroism (CD) was also proposed.39 The method relies on the properties of a particular aza dye, the diethylthiadicarbocyanine dye [DiSC2(5)], which has a strong tendency to aggregate on PNA–DNA duplexes. This aggregation gives rise to a characteristic band in the visible spectrum at 540 nm, with the appearance of a typical purple color. This feature, which could potentially lead to the development of colorimetric tests for directly visualizing the specific PNA–DNA interaction, is nevertheless made less specific from the tendency of the DiSC2(5) dye to aggregate even on free PNA molecules, introducing a strong bias to the colorimetric analysis which can lead to many false positives, especially when excess PNA is to be used, as it is frequent in the case of post-PCR confirmative analyses.40 However, when observed by spectropolarimetry, the dye aggregate on the PNA–DNA duplex gives rise to a very strong exciton coupling effect at the same wavelength, which could be easily visualized by CD, since the helical chirality of the duplex is transferred to the dye aggregate. The dye being achiral, the free dye in solution does not give rise to any CD signal and, what is most interesting for the robustness of the method, even the dye-free PNA aggregate is “invisible” when observed by the CD technique, standard PNAs also being non-chiral molecules. The method was first optimized with oligonucleotides, then applied to the identification of DNA extracted and amplified by PCR from peanuts, peanut-containing and peanut-free foods, allowing for a very sensitive detection. Typical results are shown in Fig. 9. The PNA recognizes and binds the DNA amplified from pure peanuts, inducing the dye aggregation on the PNA–DNA duplex which gives rise to an intense exciton coupling effect. Also the peanut DNA present in cereal snacks is easily detectable, albeit with a lower signal, by this method. A chocolate wafer without peanut gave no CD signal: in the absence of any PNA–DNA duplex, the dye aggregation does not take place nor is visible on the free PNA.39
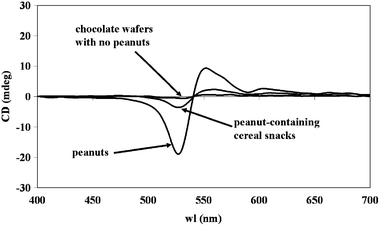 | ||
| Fig. 9 A CD-based experiment with a PNA probe specific for peanut DNA together with Disc2(5) dye for the post-PCR confirmation of the DNA identity (reproduced with permission from ref. 39). | ||
4. Microbial contaminant detection
The identification of microbial contaminants is a primary issue for food safety. As a typical example, among the 2008 data concerning the total alerts in food and feed, as included in the Rapid Alert System, 24% were about potentially pathogenic microorganisms, the highest relative category of risk.41 The ideal analysis for assessing food microbial contaminants should be fast, sensitive and selective, in order to avoid false positive and false negative results and to yield data on the eventual contamination in the shortest possible time.In order to respond to these issues, several methods for rapid microbiology based on PNA probes appeared in the last years and several examples of detection of microbial agents having food relevance were also reported.42 One of the mostly used methods is PNA-based Fluorescence In situ Hybridization (PNA-FISH). In this assay fluorophore-labelled PNAs targeted to rRNA are used for the direct detection of microorganisms in tissues, biological fluids, culture media, filters, on slides or in solution.
The advantages of PNA-FISH methods, rather than the more common DNA-FISH, were outlined in a work aimed at developing PNA probes targeted at the detection of whole cells of Listeria spp. (Fig. 10).43PNA probes turned out to be more able than DNA probes to penetrate recalcitrant biological structures such as the membranes of gram-positive cells. Moreover, due to their high affinity for complementary RNA sequences, combined to the independence from ionic strength of PNA–RNA stability, PNAs were able to bind regions on the ribosomes which were inaccessible for DNA probes, exploiting hybridization conditions which can denature RNA. Actually, when using DNA probes, any attempt of destabilizing RNA secondary structures on the target unavoidably yields also a loss of the probe affinity for the target itself; in contrast, with PNA probes, as in the reported example, recognition took place under low salt, high temperature and high pH conditions, in which RNA is likely to be in a non-native form.
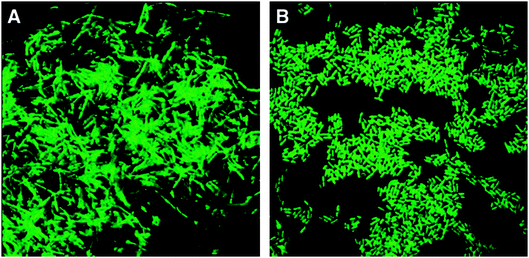 | ||
| Fig. 10 Typical PNA hybridization results in PNA-FISH applications. (A) Cells of Bacillus cereus ATCC 11778 hybridized with the universal bacterial PNA probe. (B) Cells of Listeria monocytogenesFSL-C1-122 hybridized with the Listeria-specific PNA probe (reproduced with permission from ref. 43). | ||
In a similar example, PNA probes developed to detect Campylobacter jejuni, Campylobacter coli and Campylobacter lari were able to detect C. coli spiked in drinking water samples, after membrane filtration to concentrate the microorganisms.44 A PNA-FISH procedure targeted to Acinetobacter spp. and Pseudomonas aeruginosa was recently applied for detection, after fixation on slides, showing 100% specificity and 100% selectivity towards the former and 100% specificity and 95% selectivity towards the latter.45 A commercial version of PNA-FISH was developed in order to detect both gram-negative and gram-positive bacteria. In a particular striking example four differently labelled PNA probes were used for the simultaneous detection ofEscherichia coli, Salmonella enterica, P. aeruginosa and Staphylococcus aureus.42 In a similar variant, named PNA Chemiluminescent In situ Hybridization (PNA-CISH), PNAs labelled with soybean peroxidase are hybridized with the target and then treated with a chemiluminescent substrate, generating light captured by a camera system. Combination of this system with previous membrane filtration, in order to concentrate the microorganisms to be detected, allowed detection ofP. aeruginosa in bottled water, E. coli in tap water and Dekkera bruxellensis in wine.42
5. Ingredient authentication
The authenticity of food products is an important issue which is recently gaining increasing attention: correct labelling and traceability of the ingredients through all stages of production, processing, and distribution has become of primary importance in many western countries. Among many labelling declarations which claim “quality” characteristics of a given food, and often difficult to be proved objectively, most concern varieties of vegetables or particular breeds of animals used as ingredients. Typical examples include, just to name a few, cheeses made only from sheep milk, wines produced from defined grape varieties, monocultivar olive oils, minced meat or fish declared from a given breed, and so on. DNA markers are well suited for traceability purposes, due to the remarkable persistence of DNA, even in the hostile environments found during many processing steps used for food production. The use of DNA markers as diagnostic tools for ingredient authenticity in food matrices has been investigated in an increasing number of projects worldwide.46 However, some processed foods contain highly degraded DNA which may affect the subsequent PCR used for the amplification of the diagnostic DNA sequence. In these cases, a very sensitive and specific method for the detection of small amounts of DNA has become therefore highly desirable. Even if the literature in the field is still quite scarce, it is obvious that all the PNA-based techniques so far described might be very useful in order to assess food authenticity and avoid frauds. As an example, the authors assayed the presence of hazelnut oil in extra virgin olive oil by using the PNA microarray system already reported for the detection of hazelnut as a hidden allergen.38 In a preliminary test model, down to 5% hazelnut oil in extra virgin olive oil could be detected by this method (Fig. 11).47 Hazelnut olive oil may be used to adulterate extra virgin olive oil on account of the similarity of the lipophilic components, which prevent the discrimination. In this case, the adulteration is not only a fraud, but can be dangerous to people allergic to hazelnuts.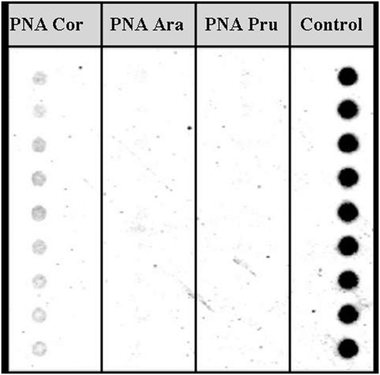 | ||
| Fig. 11 PNA microarray designed for allergen detection (hazelnut, peanut, almond) after hybridization with hazelnut DNA extracted and amplified from extra virgin olive oil spiked with 5% hazelnut oil (from ref. 47). | ||
Recently, in a mass spectrometry-based approach, modified pyrrolidinyl-PNA probes were used for targeting DNA after extraction and amplification from food samples. The PNA probes were mixed with the PCR products obtained from food and other matrices and allowed to hybridize to the eventual complementary DNA sequences. By using an ion-exchange capture technique, the DNA molecules, either free or bound to PNA, were then captured on a strong anion exchanger. After washing, MALDI-TOF analysis of the captured material targeted at detecting PNA molecules allowed to confirm the presence or the absence of PNAs, thus confirming the presence or the absence of the target DNA strand (since the PNA probes could be present only if captured together by their complementary sequences). This assay was used for detecting Single Nucleotide Polymorphisms (SNPs) in the target DNA sequence and was specific and sensitive enough to carry out simultaneous multiplex SNP detection with multiple PNA probes. The analysis could be carried out at room temperature without the need for enzyme treatment or heating. In a proof-of-concept example, different meat species in feedstuffs were identified.48PNAs are particularly useful for detecting SNPs, and thus different vegetal varieties or animal breeds, on account of their extremely high binding sequence selectivity.
6. PNA-based analyses in foods: trends and future perspectives
The above reported examples have outlined the tremendous potential that PNA-based technologies have in order to perform many different types of food analyses, such as microbial contamination, the presence of GMOs or of hidden allergens or ingredient authentication. The main methods presented in this review are summarized in Table 1.| Target DNA/RNA | Detection method | PNA | PCR | DNA labelling | Detection limit | Ref. |
|---|---|---|---|---|---|---|
| a Not reported. | ||||||
| GM soy/maize DNA | PCR clamping | Standard | Yes | No | n.r.a | 21 |
| GM maize DNA | Real time immuno-PCR | Surface | Yes | Biotin | 6 amol | 22 |
| GM soy DNA | HPLC-fluorescence | Standard | Yes | Cy5 | 200 fmol | 24 |
| GM soy DNA | HPLC-fluorescence | Beacon | Yes | No | n.r.a | 26 |
| GM soy/maize DNA | Microarray | Surface | Yes | Cy5 | 65 fmol | 29 and 30 |
| GM soy DNA | SPR | Surface | Yes | No | n.r.a | 31 |
| GM soy DNA | SPR imaging | Surface | No | No | 18 ymol | 34 |
| Peanut DNA | HPLC-fluorescence | Standard | Yes | Cy5 | n.r.a | 37 |
| Hazelnut/peanut DNA | Microarray | Surface | Yes | Cy5 | 65 fmol | 38 |
| Peanut DNA | Circular dichroism | Standard | Yes | No | 10 pmol | 39 |
| Bacterial RNA | Multiplex FISH | Cy5, Cy3, DEAC, fluorescein | No | No | n.r.a | 42 |
| Listeria RNA | FISH | Fluorescein | No | No | n.r.a | 43 |
| Campylobacter RNA | FISH | TAMRA | No | No | n.r.a | 44 |
| Animal DNA in meat | MALDI-TOF | Standard | Yes | No | n.r.a | 48 |
PNA probes have several advantages which bypass many problems often encountered when using oligonucleotide probes: higher affinity and selectivity towards the complementary DNA sequences, outstanding chemical and biological stability, higher independence of hybridization from environment conditions (pH, ionic strength), easiness of functionalization in order to obtain the desired chemical, physical or biological characteristics.
The selectivity of the probes can be further increased by using modified PNA monomers.49 One of the most efficient modifications was the introduction of PNA monomers derived from chiral amino acids with positively charged side chains (chiral PNAs),50 which were shown to be selective in recognition of single point mutations with different techniques.51
Obviously, in every assay, the PNA role is to recognize a particular DNA or RNA sequence. A crucial point for the development of any application is to find a way to transduce the hybridization events into suitable signals, strong enough to be detected. Also from this point of view, PNAs were shown to be very flexible probes, which have been used with many different techniques, ranging from very sophisticate to quite simple ones: microfluidic, microarray, capillary electrophoresis, liquid chromatography, gel electrophoresis, colorimetric assays. Many are the detection techniques so far used for identifying the hybridization events, and thus the presence of a particular sequence: surface plasmon resonance, circular dichroism, UV spectrophotometry, fluorescence, electrochemical properties, or even naked eye. In particular, advanced sensor technologies can benefit from the use of PNA probes in combination with different detection methods. The capture of a negatively charged DNA sequence by a neutral PNA probe deposited on the surface of an electrode (PNA can be adsorbed onto carbon electrodes) induces a dramatic change in the potential, which is transformed into an electrochemical signal;52,53 miniaturized sensing systems can be achieved using impedance measurements on surfaces modified with PNAs;54,55 mechanical detection can be achieved by a change in the frequency of quartz crystal microbalance (QCM),56 but future applications can also be envisaged in the use of micro- and nanocantilevers.57 All the above mentioned techniques have greatly benefited by the use of PNA probes in terms of sensitivity and selectivity.
On account of the high flexibility, PNAs open wide possibilities in the field of DNA detection in foods. Thus, it is to be expected that they will be more and more useful tools in food analysis and food authentication. In this field what is often needed can be summarized in two words: fast and reliable. In this context a likely evolution of the current state of the art is in the direction of making the plethora of already existing PNA-based assays simpler and more robust.
In particular, a likely evolution, not only in the PNA field, but concerning DNA analysis in general, is the possibility to detect DNA without any need of labelling it (label-free) or even, in the most advanced applications, without pre-amplification by PCR (PCR-free), a technique often prone to many bias and errors. In this case it is not incorrect to state that, as far as PNAs are concerned, the future is already here: several applications presented in this review already are an example, although preliminary, of label-free and PCR-free DNA detection (Table 1). Techniques which are promising for the label-free detection of DNA are those based on plasmonics, such as surface plasmon resonance (SPR) techniques58,59 and photonics60 (i.e. read out of the properties of an electromagnetic field confined in a microstructured medium as a result of the events occurring at the interfaces), using optical devices such as waveguides or photonic crystal fibers,61 but also clever examples of PCR-free colorimetric detection of bacterial DNA with enzymatic assays using suitably derivatized PNAs have already been preliminarily presented in the literature.62
What is now needed is the implementation of these first steps to routinary DNA detection techniques, since PNAs have already been proven to be well suited to the task.
Acknowledgements
Italian Ministry of Education, University and Research (MIUR) is gratefully acknowledged for fundings through the Projects of Relevant National Interest 2007 scheme (PRIN 2007, contract number 2007F9TWKE)References
- M. Hernandez, D. Rodriguez-Lazaro and A. Ferrando, Curr. Anal. Chem., 2005, 1, 203–221 CrossRef CAS.
- A. Lauri and P. O. Mariani, Genes Nutr., 2009, 4, 1–12 Search PubMed.
- R. E. Poms, E. Anklam and M. Kuhn, J. AOAC Int., 2004, 87, 1391–1397 CAS.
- G. Di Bernardo, S. Del Gaudio, U. Galderisi, A. Cascino and M. Cipollaro, Biotechnol. Prog., 2007, 23, 297–301 CrossRef CAS.
- PCR Methods in Foods, ed. J. Maurer, Springer, Berlin, Germany, 2006 Search PubMed.
- F. Weighardt, Nat. Biotechnol., 2007, 25, 1213–1214 CrossRef CAS.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CrossRef CAS.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 566–568 CrossRef CAS.
- (a) B. Ren, J. M. Zhou and M. Komiyama, Nucleic Acids Res., 2004, 32, 42e CrossRef; (b) B. S. Gaylord, M. R. Massie, S. C. Feinstein and G. C. Bazan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 34–39 CrossRef CAS.
- L. S. Yilmaz, H. E. Okten and D. R. Noguera, Appl. Environ. Microbiol., 2006, 72, 733–744 CrossRef CAS.
- V. A. Demidov, V. N. Potaman, M. D. Frank-Kamenetskii, M. Egholm, O. Buchardt, S. H. Sonnichsen and P. E. Nielsen, Biochem. Pharmacol., 1994, 48, 1310–1313 CrossRef CAS.
- N. Sahu, G. Shilakari, A. Nayak and D. H. Kohli, Curr. Chem. Biol., 2008, 2, 110–121 Search PubMed.
- P. E. Nielsen and M. Egholm, Peptide Nucleic Acids: Protocols and Applications, Horizon Press, Wymondham, Norfolk, UK, 2nd edn, 2004 Search PubMed.
- (a) H. Orum, P. E. Nielsen, M. Egholm, R. H. Berg, O. Buchardt and C. Stanley, Nucleic Acids Res., 1993, 21, 5332–5336 CAS; (b) H. Orum, Curr. Issues Mol. Biol., 2000, 2, 27–30 Search PubMed.
- L. Cerqueira, N. F. Azevedo, C. Almeida, T. Jardim, C. W. Keevil and M. J. Vieira, Int. J. Mol. Sci., 2008, 9, 1944–1960 Search PubMed.
- C. Tapparel, S. Cordey, S. Van Belle, L. Turin, W. M. Lee, N. Regamey, P. Meylan, K. Mühlemann, F. Gobbini and L. Kaiser, J. Clin. Microbiol., 2009, 47, 1742–1749 CrossRef CAS.
- O. Brandt and J. D. Hoheisel, Trends Biotechnol., 2004, 22, 617–622 CrossRef CAS.
- (a) Regulation (EC) No 1829/2003 The European Parliament and the Council of the European Union on genetically modified food and feed. Off. J. Eur. Union 2003, L268, 1–23; (b) The European Parliament and the Council of the European Union Regulation (EC) No 1830/2003 concerning the traceability and labelling of genetically modified organisms and the traceability of food and feed products produced from genetically modified organisms and amending Directive 2001/18/EC. Off. J. Eur. Union 2003, L268, 24–28; (c) Commission Regulation (EC) No 65/2004 establishing a system for the development and assignment of unique identifiers for genetically modified organisms. Off. J. Eur. Union 2004, L10, 5–10.
- Validation of methods for GMO detection is extensively carried out in the Ispra Joint Research Center of the European Commission. See http://gmo-crl.jrc.ec.europa.eu/.
- Regione Piemonte, Ordinanza Presidente Giunta Regionale n. 63, 11/7/2003; Regione Lombardia DGR13848/2003.
- C. Peano, F. Lesignoli, M. Gulli, R. Corradini, M. C. Samson, R. Marchelli and N. Marmiroli, Anal. Biochem., 2005, 344, 174–182 CrossRef CAS.
- L. Roth, J. Zagon, A. Ehlers, L. W. Kroh and H. Broll, Anal. Bioanal. Chem., 2009, 394, 529–537 CrossRef CAS.
- F. Lesignoli, A. Germini, R. Corradini, S. Sforza, G. Galaverna, A. Dossena and R. Marchelli, J. Chromatogr., A, 2001, 922, 177–185 CrossRef CAS.
- S. Rossi, F. Lesignoli, A. Germini, A. Faccini, S. Sforza, R. Corradini and R. Marchelli, J. Agric. Food Chem., 2007, 55, 2509–2516 CrossRef CAS.
- A. Castro and J. G. K. Williams, Anal. Chem., 1997, 69, 3915–3920 CrossRef CAS.
- F. Totsingan, S. Rossi, R. Corradini, T. Tedeschi, S. Sforza, A. Juris and E. Scaravelli, Org. Biomol. Chem., 2008, 6, 1232–1237 RSC.
- F. Totsingan, T. Tedeschi, S. Sforza, R. Corradini and R. Marchelli, Chirality, 2009, 21, 245–253 CrossRef CAS.
- A. Germini, A. Zanetti, C. Salati, S. Rossi, C. Forre, S. Schmid, C. Fogher and R. Marchelli, J. Agric. Food Chem., 2004, 52, 3275–3280 CrossRef.
- A. Germini, A. Mezzelani, F. Lesignoli, R. Corradini, R. Marchelli, R. Bordoni, C. Consolandi and G. De Bellis, J. Agric. Food Chem., 2004, 52, 4535–4540 CrossRef CAS.
- A. Germini, S. Rossi, A. Zanetti, R. Corradini, C. Fogher and R. Marchelli, J. Agric. Food Chem., 2005, 53, 3958–3962 CrossRef CAS.
- H. Park, A. Germini, S. Sforza, R. Corradini, R. Marchelli and W. Knoll, Biointerphases, 2007, 2, 80–88 CrossRef CAS.
- H. Park, A. Germini, S. Sforza, R. Corradini, R. Marchelli and W. Knoll, Biointerphases, 2006, 1, 113–122 CrossRef CAS.
- R. D'Agata, R. Corradini, G. Grasso, R. Marchelli and G. Spoto, ChemBioChem, 2008, 9, 2067–2070 CrossRef CAS.
- R. D'Agata, R. Corradini, C. Ferretti, L. Zanoli, M. Gatti, R. Marchelli and G. Spoto, Biosens. Bioelectron., 2010, 25, 2095–2100 CrossRef CAS.
- S. Cochrane, K. Beyer, M. Clausen, M. Wjst, R. Hiller, C. Nicoletti, Z. Szepfalusi, H. Savelkoul, H. Breiteneder, Y. Manios, R. Crittenden and P. Burney, Allergy (Oxford, U. K.), 2009, 64, 1246–1265 Search PubMed.
- A. van Hengel, Anal. Bioanal. Chem., 2007, 389, 111–118 CrossRef CAS.
- A. Germini, E. Scaravelli, F. Lesignoli, S. Sforza, R. Corradini and R. Marchelli, Eur. Food Res. Technol., 2005, 220, 619–624 CrossRef CAS.
- S. Rossi, E. Scaravelli, A. Germini, R. Corradini, C. Fogher and R. Marchelli, Eur. Food Res. Technol., 2006, 223, 1–6 CrossRef CAS.
- S. Sforza, E. Scaravelli, R. Corradini and R. Marchelli, Chirality, 2005, 17, 515–521 CrossRef CAS.
- T. Tedeschi, S. Sforza, S. Ye, R. Corradini, A. Dossena, M. Komiyama and R. Marchelli, J. Biochem. Biophys. Methods, 2007, 70, 735–741 CrossRef CAS.
- The Rapid Alert System for Food and Feed (RASFF), Annual Report 2008, European Commission.
- (a) H. Perry-O'Keefe and J. J. Hyldig-Nielsen, in Environmental: Molecular Microbiology Protocols and Applications, ed. P. Rochelle, Horizon Scientific Press, Wymondham, UK, 2001 Search PubMed; (b) H. Stender, M. Fiandaca, J. J. Hyldig-Nielsen and J. Coull, J. Microbiol. Methods, 2002, 48, 1–17 CrossRef CAS.
- B. F. Brehm-Stecher, J. J. Hyldig-Nielsen and E. A. Johnson, Appl. Environ. Microbiol., 2005, 71, 5451–5457 CrossRef CAS.
- M. J. Lehtola, C. J. Loades and C. W. Keevil, J. Microbiol. Methods, 2005, 62, 211–219 CrossRef CAS.
- A. Y. Peleg, Y. Tilahun, M. J. Fiandaca, E. M. C. D'Agata, L. Venkataraman, R. C. Moellering Jr and G. M. Eliopoulos, J. Clin. Microbiol., 2009, 47, 830–832 CrossRef CAS.
- S. Doveri and D. Lee, J. Agric. Food Chem., 2007, 55, 4640–4644 CrossRef CAS.
- S. Rossi, PhD thesis, University of Parma, 2007.
- B. Boontha, J. Nakkuntod, N. Hirankarn, P. Chaumpluk and T. Vilaivan, Anal. Chem., 2008, 80, 8178–8186 CrossRef CAS.
- K. N. Ganesh and P. E. Nielsen, Curr. Org. Chem., 2000, 4, 931–943 CAS.
- (a) S. Sforza, R. Corradini, S. Ghirardi, A. Dossena and R. Marchelli, Eur. J. Org. Chem., 2000, 2905–2913 CrossRef CAS; (b) V. Menchise, G. De Simone, T. Tedeschi, R. Corradini, S. Sforza, R. Marchelli, D. Capasso, M. Saviano and C. Pedone, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12021–12026 CrossRef CAS.
- (a) R. Corradini, G. Feriotto, S. Sforza, R. Marchelli and R. Gambari, J. Mol. Recognit., 2004, 17, 76–84 CrossRef CAS; (b) M. Chiari, G. Galaverna, S. Sforza, M. Cretich, R. Corradini and R. Marchelli, Electrophoresis, 2005, 26, 4310–4316 CrossRef CAS.
- J. Wang, Biosens. Bioelectron., 1998, 13, 757–762 CrossRef CAS.
- P. Kara, K. Kerman, D. Ozkan, B. Meric, A. Erdem, P. E. Nielsen and M. Ozsoz, Bioelectrochemistry, 2002, 58, 119–126 CrossRef.
- A. Macanovic, Nucleic Acids Res., 2004, 32, 20e CrossRef.
- J. Liu, S. Tian, P. E. Nielsen and W. Knoll, Chem. Commun., 2005, 2969–2971 RSC.
- J. Wang, P. E. Nielsen, M. Jiang, X. Cai, J. R. Fernandes, D. H. Grant, M. Ozsoz, A. Beglieter and M. Mowat, Anal. Chem., 1997, 69, 5200–5202 CrossRef CAS.
- K. S. Hwang, S. M. Lee, S. K. Kim, J. H. Lee and T. S. Kim, Annu. Rev. Anal. Chem., 2009, 2, 77–98 Search PubMed.
- C. Ananthanawat, T. Vilaivan, V. P. Hoven and X. Su, Biosens. Bioelectron., 2010, 25, 1064–1069 CrossRef CAS.
- J. Homola, Chem. Rev., 2008, 108, 462–493 CrossRef CAS.
- X. Fan, I. M. White, S. I. Shopova, H. Zhu, J. D. Suter and Y. Sun, Anal. Chim. Acta, 2008, 620, 8–26 CrossRef CAS.
- (a) F. Poli, A. Cucinotta and S. Selleri, Photonic crystal fibers, in Material Science, Springer-Verlag, Dordrecht, The Netherlands, vol. 102 , 2007 Search PubMed; (b) A. Coscelli, M. Sozzi, F. Poli, D. E. Passaro, A. Cucinotta, S. Selleri, R. Corradini and R. Marchelli, IEEE J. Sel. Top. Quantum Electron., 2010, 16, 967–972 CrossRef.
- N. Zhang and D. H. Appella, J. Am. Chem. Soc., 2007, 129, 8424–8425 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |