Construction of three high-dimensional supramolecular networks from temperature-driven conformational isomers†
Hua-Cai
Fang
,
Ying-Ying
Ge
,
Hong-Yang
Jia
,
Shan-Shan
Li
,
Feng
Sun
,
Li-Guo
Zhang
and
Yue-Peng
Cai
*
School of Chemistry and Environment, Key Lab of Technology on Electrochemical Energy Storage and Power Generation in Guangdong Universities, Engineering Research Center of Materials and Technology for Electrochemical Energy Storage (Ministry of Education), South China Normal University, Guangzhou, 510006, China
First published on 1st November 2010
Abstract
Under different temperatures, the reaction mixture of the multidentate organic ligand usy-L4 and Zn(NO3)2 + NaN3 crystallized to give three conformational isomers, namely Zn(usy-L4)(η1-N3)2 (α-1, β-2 and γ-3) (usy-L4 = N,N-dimethyl-2-(pyridin-2-ylmethylimino)ethanamine). Single-crystal X-ray diffraction analyses reveal that the conformational isomerism of three compounds 1–3 stems from the different orientations of two azide groups with η1-terminal nitrogen atom coordinating to central zinc ion in reference to the basal chelating plane of ligand usy-L4, and further resulting in different supramolecular networks with two-dimensional 44 rhombic-grid for α-1, a three-dimensional 36·418·53·6 compressed-NbO framework for β-2, and a three-dimensional “dense” 424·64 topology for γ-3. Meanwhile, three isomers may be also irreversibly converted from 1 to 2, to 3 through SC-to-SC transformation driven by temperature.
The current interest in the crystal engineering of coordination complexes stems from their potential applications as materials for gas storage, separation, and catalysis, as well as their intriguing variety of topologies.1 The azide ion, N3−, is a versatile ligand2 that can coordinate to metal ions by a variety of modes and conformations, thus making it not only an interesting exchange coupler for paramagnetic metal ions in the field of molecular magnetism,3 but also a very useful linker (as acceptor of hydrogen bonds) for donor of hydrogen bonds, that is C(O,N,S)-H, in the supramolecular system.4 Accordingly, the self-assembly of multidentate organic ligands together with azide, N3−, and metal ions has resulted in many supramolecular networks constructed by coordination or/and hydrogen bonds or other weaker supramolecular interactions, such as π⋯π stacking interactions.5 Although concepts of crystal engineering and supramolecular chemistry are usefully employed in the attempt to construct networks of desired topologies and properties, many subtle factors, for example, the selection of the metal ions with different coordination geometry or radius, counteranions with different coordination abilities, or bulk, solvent, metal/ligand ratio, and even pH conditions still contribute to make this objective a major challenge.2c,6 For these influencing factors, the reaction/crystallization temperature and different orientations of the auxiliary ligand, in particular, play a very important role in the self-assembly processes of metal-complexes with different structural topologies.
On the other hand, tridentate N2O and tetradentate N2O2 ligands such as acacen-/pyridine/salen-type Schiff bases7,8 (ESI, Scheme S1)† are capable of forming complexes with certain metal ions, which exhibit unusual coordination and potentially useful chemotherapeutic properties.9 In recent years, our research group has already reported a series of transition or lanthanide metal complexes involving symmetrical or asymmetrical Schiff base ligands with N2O, N2O2, and N2S donor sets, for example, mono- or multinuclear copper(II) and zinc(II) complexes containing the symmetrical tetradentate Schiff bases bis(acetylacetone)trimethylene-diimine (H2acactn, sy-H2L1) and bis(salicylidene)trimethylenediamine (sy-H2L3).10 Meanwhile, three novel μ-hydroxo-bridged tetranuclear organolanthanide complexes with bis(acetylacetone)ethylene-diamine (H2acacen = sy-H2L2) and sy-H2L3 were assembled under a N2 atmosphere and anhydrous conditions.11 Recently, a Zn(II) complex involving the ligand sy-H3L2 with selective fluorescent chemosensor for Ni2+ ion was reported.12a Simultaneously, some novel mono- and trinuclear Cd(II) complexes with the tripodal/dipolar imine-phenol Schiff base ligands sy-H3L4 and sy-H2L5 through the different strategies of synthesis were also assembled.12b In particular, very recently we have prepared two tridentate unsymmetrical Schiff base ligands 2-((2-(dimethylamino)ethylimino)-methyl)phenol (usy-HL1) and methyl-2-pyridylmethyl-idenehy-drazinecarbodithioate(usy-HL3), through the different synthetic strategies, several mononuclear Zn(II), Co(II, III), Mn(II), Ni(II) compounds and a series of anion-/pH-controlled low-dimensional cadmium(II) compounds were constructed.13
As part of our ongoing study of metal complexes of the multifunctional Schiff bases, we report here under different temperatures, the reaction mixture of the multidentate organic ligand usy-L4 and Zn(NO3)2 + NaN3 crystallized to give three zinc conformational isomers, namely Zn(usy-L4)(N3)2 (α-1, β-2 and γ-3) (usy-L4 = N,N-dimethyl-2-(pyridin-2-ylmethylimino)ethanamine), and investigate that the different orientations of two azide groups with η1-terminal nitrogen atom coordinating to central zinc ion in reference to the basal chelating plane of lignad usy-L4 affect on the supramolecular structures of three conformational isomers 1–3. Meanwhile, SC-to-SC irreversible transformations between the three isomers from 1 to 2, to 3 are also studied. In contrast to the aforementioned cases containing auxiliary ligand of azide, fewer efforts have been devoted to the systematic investigation of azide coordination chemistry for influencing the supramolecular networks of the reaction outcomes.
The three compounds 1–3 were obtained by the reaction of ligand usy-L4 with NaN3 and Zn(NO3)2 in methanol solution for three hours, and crystallized at different temperatures with −5 °C for compound 1, 6 °C for compound 2 and 26 °C for compound 3 (Scheme 1).‡
 | ||
| Scheme 1 Syntheses and conversation of three conformational isomers α-1, β-2 and γ-3. | ||
The structures of the three complexes were identified by elemental analysis, IR and X-ray diffraction. The IR spectra of usy-L4 and the corresponding complexes 1–3 are very similar and provide information about the metal–ligand bonding. The assignments are based on the typical group frequencies. A weak broad band in the region 3565 cm−1 or so due to a hydrogen bonded C–H⋯N interactions in three compounds 1–3. The strong ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) bands occurring in the range of 1597–1635 cm−1 for these complexes are shifted considerably toward lower frequencies compared to that of the free Schiff base ligand usy-L4 (1646 cm−1), showing that the azomethine nitrogen atom is coordinated to the metal.12 The weak band in the region of 462–467 cm−1 for the three complexes can be assigned to ν(M–N),12 and provides further evidence for coordination through the terminal nitrogen atoms. The position of the 2063 cm−1 (νas-N3) falls within the absorption range of terminal azides in transition metal complexes,14 and the appearance of the νs-N3 band at 1341 cm−1 indicates the asymmetric nature of the azide groups in three compounds 1–3.
N) bands occurring in the range of 1597–1635 cm−1 for these complexes are shifted considerably toward lower frequencies compared to that of the free Schiff base ligand usy-L4 (1646 cm−1), showing that the azomethine nitrogen atom is coordinated to the metal.12 The weak band in the region of 462–467 cm−1 for the three complexes can be assigned to ν(M–N),12 and provides further evidence for coordination through the terminal nitrogen atoms. The position of the 2063 cm−1 (νas-N3) falls within the absorption range of terminal azides in transition metal complexes,14 and the appearance of the νs-N3 band at 1341 cm−1 indicates the asymmetric nature of the azide groups in three compounds 1–3.
The structural analysis reveals that three complexes 1–3 are conformational isomers with the same composition [Zn(usy-L4)(η1-N3)2] (α-1, β-2 and γ-3), and their molecular structures are shown in Fig. 1 and Fig. S1–2,† respectively. The important bond lengths for three compounds are listed in Fig. 1 and Fig. S1–2.† The atom-numbering scheme adopted is the same for three compounds. Each complex is neutral mononuclear, in which the Zn(II) is five-coordinated with N5 donor set by three nitrogen atoms from the same Schiff base ligand usy-L4 and other two ones from two azide ions. The Zn–N distances from 1.977(2) to 2.263(2) for α-1, 1.977(3) to 2.275(3) for β-2 and 1.974(4) to 2,250(4) Å for γ-3, and the angles N–Zn–N around Zn2+ are 74.89(14) to 158.31(13)° for α-1, for 75.17(11) to 152.63(12)° β-2 and 74.63(7) to 151.25(8)° for γ-3, and the coordination geometry around the zinc is best regarded as a distorted trigonal bipyramid with the two nitrogen atom (N1, N3) occupying the apical position and N2, N4, N7 defining the basal plane as indicated in Fig. 1 and Fig. S1–2.†
 | ||
| Fig. 1 Molecular structure of isomer α-1 with the atom numbers. The selected bond distances Zn1–N1 2.264(3), Zn1–N2 2.083(3), Zn1–N3 2.230(3), Zn1–N4 1.978(3), Zn1–N7 1.984(3) Å. | ||
Compared with three conformational isomers, the main difference is from different space orientations of two coordinated azide ions in their compounds, namely, two azide ions leave the paper in α-1 and point to the paper in β-2, as well as one points to the paper and another leaves the paper in γ-3 as shown in Scheme 2. Based on these differences, through intermolecular hydrogen bonding C–H⋯N C−1–H⋯π and π⋯π packing interactions, the different supramolecular structures were obtained respectively (Schemes 1–2 and Fig. S3–5).†
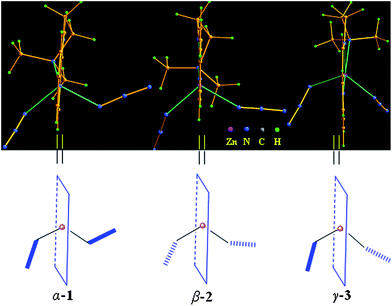 | ||
| Scheme 2 Orientation of two coordinated azide ions in reference to the basal chelating plane of ligand usy-L4 in three conformational isomers α-1, β-2 and γ-3. | ||
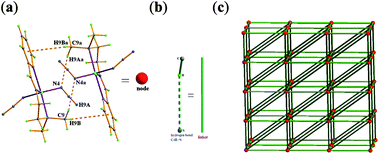
In compound 1, via intermolecular hydrogen bonding C–H⋯N and π⋯π packing interactions (ESI, Table S1),† the dimmer unit is formed as shown in Fig. 2. Each neutral dimmer unit forms a series of acceptor/donor hydrogen bonds with four neighbouring ones via intermolecular hydrogen bond involving C–H⋯N, leading to the formation of two-dimensional ordered layer-like structure (ESI, Fig. S3).† If the dimmer units are viewed as the nodes (Fig. 2a) and the hydrogen bonding C–H⋯N interactions from four neighbouring ones as linkers (Fig. 2b), in the case, the resulted 2D supramolecular structure (ESI, Fig. S3)† may be simplified into two-dimensional 44 rhombic-grid for isomer α-1 as depicted in Fig. 2c.
 | ||
| Fig. 2 Two-dimensional 44 rhombic-grid for α-1. | ||
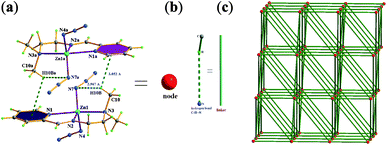
As for isomer β-2, the dimmer unit as the node of network is formed viahydrogen bonding C–H⋯N and C–H⋯π interactions as depicted in Fig. 3a, and is similar to isomer α-1, the linker in isomer β-2 is still intermolecular hydrogen bonding C–H⋯N interactions (Fig. 3b). Because each neutral dimmer unit (node) is connected to eight adjacent nodes via these linkers (C–H⋯N interactions), in this case, resulting in the formation of a three-dimensional 36·418·53·6 compressed-NbO framework for isomer β-2 as shown in Fig. 3c.
 | ||
| Fig. 3 3D 36·418·53·6 compressed-NbO framework for isomer β-2. | ||
The packing structure of compound 3 is shown in Fig. 4. By closely observing Fig. 2 and 4, it is found that there are nodes and linkers similar to β-2, however the final 3D supramolecular network is different with a “dense” 424·64 topology in isomer γ-3 (Fig. 4c) and 36·418·53·6 compressed-NbO one for β-2 (Fig. 3c), mainly deriving from the different space orientations of two coordinated η1-terminal azide ions.
 | ||
| Fig. 4 The 3D “dense” 424·64 topology for γ-3. | ||
Another important feature of the crystals of molecules 1–3 is the irreversible single-crystal to single-crystal transformation driven by temperature. From the molecular structures of discrete 1–3 (Fig. 1 and Fig. S1–2),† we find that two coordinated azide ions are η1-terminal mode and have some conformational flexibility around the N–Zn bonds, thus providing favourable conditions for a topochemical reaction. When crystals of isomers α-1 and β-2 were heated from room temperature to 40–55 °C for 5 h under vacuum, isomers β-2 and γ-3 were obtained, respectively. Although this process is accompanied by single crystal cracking and the color changes from yellow (α-1)/pale yellow (β-2) to pale yellow (β-2)/light yellow (γ-3), the single crystallinity was still retained. Fortunately we were able to perform X-ray analysis on the smaller piece of single crystals after the transformation, which confirms that they are respective isomers β-2 and γ-3 and well consistent with the results of their EA.
The structures of the bulk materials for isomers β-2 and γ-3 were confirmed by matching their X-ray powder patterns with those generated from the corresponding single crystals (Fig. S6 in the ESI).† Transformations by thermal treatment at temperatures 40–55 °C result in the significant changes in the powder patterns, namely β-2/γ-3, but acceptable matches were observed between the simulated single-crystal X-ray data patterns and the experimental powder X-ray diffraction patterns for bulk crystalline samples as obtained from the synthesis of the corresponding compounds β-2 and γ-3. These facts clearly indicate that the phase transitions described for single crystals also occur in macroscopic powder samples and lead to monophasic products and the crystal-to-crystal transformation (SCSC) described is irreversible (ESI, Fig. S7).†
To further understand and confirm the relative thermal stability and conversion mentioned above among three conformational isomers α-1, β-2 and γ-3, their single point energy has been studied at B3LYP/6-311G(d) level by using density functional theory (DFT). As is shown in Fig. 5, single point energy for isomers α-1, β-2 and γ-3 are −1670479.1643 kJ mol−1, −1670455.0970 kJ mol−1 and −1670443.5746 kJ mol−1, respectively. Energy differences ΔE(1,2) is 24.07 kJ mol−1 and ΔE(2,3) is 11.52 kJ mol−1. The fact shows that the good agreement between the theoretical values and experimental results for three isomers, in which the different thermal stability mainly derives from different intermolecular weak interactions, such as π⋯π stacking and hydrogen bonding C–H⋯N(π) interactions.
 | ||
| Fig. 5 Single point energy of three conformational isomers 1–3 and their relative energy difference. | ||
Metal–organic frameworks have been reported to have the ability to affect the emission wavelength and intensity of the organic material through metal coordination.15 Therefore, it is important to investigate the luminescent properties of metal–organic frameworks in view of potential applications. The photoluminescent behaviour of isomers α-1, β-2, γ-3 and free ligand usy-L4 are studied in the solid state at room temperature. The emission spectra of isomers α-1, β-2, γ-3 and usy-L4 are depicted in Fig. 6. Apparently, the emission spectra of the complexes 1, 2 and 3 closely resemble that of the ligand usy-L4 excluding the emission intensity, indicating the fluorescence of the complexes 1, 2, 3 are usy-L4-based emission. Meanwhile, the blue emission for complexes 1, 2, 3 comparing with usy-L4 can be observed, where the maximum emission wavelength at 512 nm (under 404 nm excitation) for the ligand usy-L4, 453 nm (under 374 nm excitation) for complex 1, 453 nm (under 374 nm excitation) for complex 2 and 453 nm (under 374 nm excitation) for 3. Compared with the emission spectrum of usy-L4, a blue shift of 59 nm in three isomers α-1, β-2, γ-3 has been given. The incorporation of Zn(II) effectively reduces the loss of energy via vibration motions, thus the enhanced fluorescence intensities of the three complexes are detected.16
 | ||
| Fig. 6 Emission spectra of three isomers 1–3 and ligand usy-L4 in solid state at room temperature, respectively. | ||
In summary, the tridentate unsymmetric ligand, N,N-dimethyl-2-(pyridin-2-ylmethylimino)ethanamine, usy-L4, yields three mononuclear zinc complexes 1, 2 and 3 with the same composition [Zn(usy-L4)(η1-N3)2] but with two η1-terminal azide-ligands disposed in the different space orientations in reference to the basal chelating plane of ligand usy-L4, namely three conformational isomers α-1, β-2, γ-3. Due to the different intermolecular weak interactions, the three isomers have different supramolecular networks with two-dimensional 44 rhombic-grid for α-1, three-dimensional 36·418·53·6 compressed-NbO framework for β-2, and three-dimensional “dense” 424·64 topology for γ-3. Meanwhile, three isomers may be also irreversibly conversed from 1 to 2, to 3 through SC-to-SC transformation driven by temperature. Obviously, these results provide the very effective strategies for constructing different topological supramolecular networks with N2O donor tridentate asymmetrical Schiff base ligands. The strong fluorescence emission of 1, 2 and 3 make them a potentially useful photoactive materials.
Acknowledgements
The authors are grateful for the financial aids from the National Natural Science Foundation of P. R. China (grant no. 20772037 and 21071056), Science and Technology Planning Project of Guangdong Province (grant no. 2006A10902002 and 2010B031100018) and the N. S. F. of Guangdong Province (grant no. 9251063101000006 and 06025033).References
- (a) D. Maspoch, D. Ruiz-Molina and J. Veciana, Chem. Soc. Rev., 2007, 36, 770 RSC; (b) A. J. Blake, N. R. Champness, P. Hubberstey, W. S. Li, M. A. Withersby and M. Schröder, Coord. Chem. Rev., 1999, 183, 117 CrossRef CAS; (c) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS; (d) O. R. Evans and W. Lin, Acc. Chem. Res., 2002, 35, 511 CrossRef CAS; (e) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS; (f) C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466 CrossRef CAS; (g) S. Kitagawa, R. Kitaura and S. I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS; (h) U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626 RSC; (i) P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38, 2638 CrossRef.
- (a) J. Ribas, A. Escuer, M. Monfort, R. Vicente, R. Cortes, L. Lezama and T. Rojo, Coord. Chem. Rev., 1999, 193–195, 1027 CrossRef CAS and references therein; (b) L. K. Thompson and S. S. Tandon, Comments Inorg. Chem., 1996, 18, 125 CrossRef CAS; (c) P. Chaudhuri, R. Wagner, S. Khanra and T. Weyhermüller, Dalton Trans., 2006, 4962 RSC.
- M. A. Aebersold, B. Gillon, O. Plantevin, L. Pardi, O. Kahn, P. Bergerat, I. von Seggern, F. Tuczek, L. Öhrström, A. Grand and E. Leliévre-Berna, J. Am. Chem. Soc., 1998, 120, 5238 CrossRef CAS.
- (a) S. Demeshko, G. Leibeling, W. Maringgele, F. Meyder, C. Mennerich, H.-H. Klauss and H. Pritzkow, Inorg. Chem., 2005, 44, 519 CrossRef CAS; (b) Z. E. Serna, L. Lazama, M. Urtiaga, M. I. Arriortua, M. G. Barandika, R. Cortes and T. Rojo, Angew. Chem., Int. Ed., 2000, 39, 344 CrossRef CAS; (c) M. Monfort, I. Resino, J. Ribas, X. Solans, M. Font-Bardia, P. Rabu and M. Drillon, Inorg. Chem., 2000, 39, 2572 CrossRef CAS; (d) G. S. Papaefstathiou, A. Escuer, R. Vicente, M. Font-Bardia, X. Solans and S. P. Perlepes, Chem. Commun., 2001, 2414 RSC.
- (a) K. Dhara, U. C. Saha, A. Dan, S. Sarkar, M. Manasserod and P. Chattopadhyay, Chem. Commun., 2010, 46, 1754 RSC; (b) J.-Y. Zhang, Q. Yue, Q.-X. Jia, A.-L. Cheng and E.-Q. Gao, CrystEngComm, 2008, 10, 1443 RSC; (c) S. Matsumura, K. Shikano, T. Oi, N. Suzuki and H. Nagao, Inorg. Chem., 2008, 47, 9125 CrossRef CAS; (d) M. A. S. Goher, B. Sodin, B. Bitschnau, E. C. Fuchs and F. A. Mautner, Polyhedron, 2008, 27, 1423 CrossRef CAS; (e) M. Yuan, F. Zhao, W. Zhang, Z.-M. Wang and S. Gao, Inorg. Chem., 2007, 46, 11235 CrossRef CAS; (f) T. Ramachandramoorthy, A. P. Raj, V. Sivasankar and S. Rajam, Chem.–Asian J., 2007, 19, 4415–4419 CAS; (g) Angew. Chem., Int. Ed., 2006, 45, p. 3131 Search PubMed; (h) R. P. Sharma, R. Sharma, R. Bala and P. Venugopalan, J. Mol. Struct., 2006, 787, 69 CrossRef CAS.
- Z.-B. Ma, S.-B. Han, V. Ch. Kravtsov and B. Moulton, Inorg. Chim. Acta, 2010, 363, 387 CrossRef CAS.
- (a) D. X. West, A. E. Liberta, S. B. Padhye, R. C. Chikate, P. B. Sonawane, A. S. Kumbhar and R. G. Yerande, Coord. Chem. Rev., 1993, 123, 49 CrossRef CAS; (b) M. Akbar Ali and S. E. Livingstone, Coord. Chem. Rev., 1974, 13, 101 CrossRef; (c) D. X. West, S. B. Padhye and P. B. Sonawane, Struct. Bonding, 1991, 76, 1 CAS.
- (a) S. Sen, P. Talukder, S. K. Dey, S. Mitra, G. Rosair, D. L. Hughes, G. P. A. Yap, G. Pilet, V. Gramlich and T. Matsushita, Dalton Trans., 2006, 1758 RSC; (b) G. A. Morris, H. Zhou, C. L. Stern and S. T. Nguyen, Inorg. Chem., 2001, 40, 3222 CrossRef CAS; (c) H.-C. Fang, X.-Y. Yi, Z.-G. Gu, G. Zhao, Q.-Y. We, J.-Q. Zhu, A.-W. Xu and Y.-P. Cai, Cryst. Growth Des., 2009, 9, 3776 CrossRef CAS; (d) H.-C. Fang, J.-Q. Zhu, H.-Y. Jia, S.-S. Li, X. Gong, Z.-Y. Zhou, L. Chen and Y.-P. Cai, Cryst. Growth Des., 2010, 10, 3277 CrossRef CAS; (e) W.-X. Cai, Q.-W. Zhang, H. Su and Y.-L. Feng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m1572 CrossRef; (f) Z.-L. You and H.-L. Zhu, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, m397 CrossRef; (g) W.-X. Cai, Q.-W. Zhang, H. Su and Y.-L. Feng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m1572 CrossRef; (h) S. Basak, S. Sen, C. Marschner, J. Baumgartner, S. R. Batten, D. R. Turner and S. Mitra, Polyhedron, 2008, 27, 1193 CrossRef CAS; (i) Z.-L. You and H.-L. Zhu, Z. Anorg. Allg. Chem., 2006, 632, 140 CrossRef CAS.
- (a) A. E. Liberta and D. X. West, BioMetals, 1992, 5, 121 CAS; (b) D. L. Klayman, J. P. Scovill, J. Bruce and J. F. Bartosevich, J. Med. Chem., 1984, 27, 84 CrossRef CAS; (c) C. Jr. Shipman, S. H. Smith, J. C. Drach and D. L. Klayman, Antimicrob. Agents Chemother., 1981, 19, 682 CAS.
- (a) Y.-P. Cai, C.-Y. Su, A.-W. Xu, B.-S. Kang, Y.-X. Tong, H.-Q. Liu and J. Sun, Polyhedron, 2001, 20, 657 CrossRef CAS; (b) X.-X. Zhou, H.-C. Fang, Y.-Y. Ge, Z.-Y. Zhou, Z.-G. Gu, X. Gong, G. Zhao, Q.-G. Zhan, R.-H. Zeng and Y.-P. Cai, Cryst. Growth Des., 2010 Search PubMed , accepted.
- Y.-P. Cai, H.-Z. Ma, B.-S. Kang, C.-Y. Su, W. Zhang, J. Sun and Y.-L. Xiong, J. Organomet. Chem., 2001, 628, 99 CrossRef CAS.
- (a) G.-B. Li, H.-C. Fang, Y.-P. Cai, Z.-Z. Yuan, P.-K. Thallapally and J. Tian, Inorg. Chem., 2010 Search PubMed accepted; (b) X.-X. Zhou, Y.-P. Cai, S.-Z. Zhu, Q.-G. Zhan, M.-S. Liu, Z.-Y. Zhou and L. Chen, Cryst. Growth Des., 2008, 8, 2076 CrossRef CAS.
- (a) J.-X. Lin, M.-L. Lin, Y.-J. Su, M.-S. Liu and Y.-P. Cai, Transition Met. Chem., 2007, 32, 338 CrossRef CAS; (b) S.-F. Hong, X.-H. Liang, H.-C. Fang, Z.-Y. Zhou, Z.-J. Hu and Y.-P. Cai, Transition Met. Chem., 2009, 34, 115 CrossRef CAS; (c) H.-C. Fang, J.-Q. Zhu, L.-J. Zhou, H.-Y. Jia, S.-S. Li, X. Gong, S.-B. Li, Y.-P. Cai, P. K. Thallapally, J. Liu and G. J. Exarhos, Cryst. Growth Des., 2010, 10, 3227; (d) H.-C. Fang, Y.-Y. Ge, Y. Ying, Z.-Y. Zhou, L. Chen, Q.-G. Zhan, S.-R. Zheng and Y.-P. Cai, CrystEngComm, 2010 10.1039/COCE00177E in press.
- (a) M. A. S. Goher, Acta Chim. Hung., 1984, 117, 215 CAS; (b) Z. Dori and R. F. Ziolo, Chem. Rev., 1973, 73, 247 CrossRef CAS; (c) J. L. Manson, A. M. Arif and J. S. Miller, Chem. Commun., 1999, 1479 RSC; (d) J. Ribas, M. Monfort, B. K. Ghosh and X. Solans, Angew. Chem., Int. Ed. Engl., 1994, 33, 2087 CrossRef; (e) X. Hao, Y. Wei and S.-W. Zhung, Chem. Commun., 2000, 2271 RSC.
- (a) G. Wu, X.-F. Wang, T. Okamura, W.-Y. Sun and N. Ueyama, Inorg. Chem., 2006, 45, 8523 CrossRef CAS; (b) D. M. Ciurtin, N. G. Pschirer, M. D. Smith, U. H. F. Bunz and H. C. zur Loye, Chem. Mater., 2001, 13, 2743 CrossRef CAS; (c) Y.-B. Dong, P. Wang, R.-Q. Huang and M. D. Smith, Inorg. Chem., 2004, 43, 4727 CrossRef CAS.
- (a) J.-G. Xu, Z.-B. Wang, Fluorescence Analytical Methods, 3rd ed; Chinese Science Publishing Company: Beijing, 2006 Search PubMed; (b) J.-Q. Chen, Y.-P. Cai, H.-C. Fang, Z.-Y. Zhou, X.-L. Zhan, G. Zhao and Z. Zhang, Cryst. Growth Des., 2009, 9, 1605 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional data. CCDC reference numbers 784672 (1), 784673 (2) and 784674 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0ce00485e |
‡ Synthesis preparation of 1–3: The mixture of 0.75 mmol Zn(NO3)2, 0.50 mmol NaN3, and 0.50 mmol of usy-L4 were added to 15 mL of methanol, and was stirred for 30 min at room temperature. After filtration, the filtrate was kept at −5 °C, 6 °C, and room temperature (26 °C) for several days to give yellow crystals of 1, pale yellow for 2, and light yellow for 3 upon slow evaporation of the solvent. The crystals were filtered and dried in air. For 1, yield (based on Zn): 57%. Found: C 32.85, H 4.23, N 34.61. Anal. calcd for C10H15N9Zn: C 33.09, H 4.14, N 34.74. IR (KBr, cm−1): ν 3565–3547 (br, s), 2996 (m), 2893 (s), 2799 (s), 2063 (m), 1634 (vs), 1596 (s), 1539 (s), 1471 (vs), 1446 (vs), 1341 (s), 1290 (vs), 1189 (vs), 1150 (s), 1021 (vs), 900 (vs), 792 (m), 765 (s), 676 (m), 639 (m), 600 (m), 571 (m), 528 (m), 462 (w). For 2, yield: 50%. Found: C 33.01, H 4.19, N 34.78. Anal. calcd for C10H15N9Zn: C 33.09, H 4.14, N 34.74. IR (KBr, cm−1): ν 3565–3543 (br, s), 2995 (m), 2889 (s), 2795 (m), 2063 (m), 1635 (vs), 1597 (s), 1539 (s), 1472 (vs), 1443 (vs), 1342 (s), 1291 (vs), 1188 (vs), 1149 (s), 1022 (vs), 903 (vs), 795 (m), 763 (s), 677 (m), 634 (m), 601 (m), 573 (m), 526 (m), 463 (w). For 3, yield: 45%. Found: C 33.19, H 4.29, N 34.59. Anal. calcd for C10H15N9Zn: C 33.09, H 4.14, N 34.74. IR (KBr, cm−1): ν 3565–3546 (br, s), 2997 (m), 2895 (s), 2797 (s), 2062 (m), 1635 (vs), 1599 (s), 1542 (s), 1473 (vs), 1449 (vs), 1338 (s), 1294 (vs), 1192 (vs), 1152 (s), 1025 (vs), 896 (vs), 795 (m), 763 (s), 679 (m), 643 (m), 598 (m), 569 (m), 528 (m), 467 (w).Conversions among 1, 2 and 3: the three compounds 1–3 can also be irreversibly transformed from 1 to 2 and 2 to 3 by a single crystal or bulk crystals of the corresponding isomers 1 and 2 (10−5 mol) were heated at 40–55 °C for 5 h under vacuum to produce a single crystal and bulk crystals of 2–3 for single-crystal X-ray diffraction and other measurements, respectively.Crystallographic studies: X-ray diffraction data were collected on a Bruker Apex II diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) at 298 K. Absorption corrections were applied by using multi-scan program SADABS. All the structures were solved by direct methods and refined with full-matrix least-squares technique using SHELXTL. All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms on organic ligands were generated by the riding mode (C–H 0.96 Å). The terminal nitrogen atom N9 of the η1-terminal coordinated azide anion in 2 was disordered into two sites with 0.5 occupancy of per position.Crystal data: For 1, C10H15N9Zn, M = 326.70, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.3950(12) Å, b = 9.5035(13) Å, c = 10.2058(14) Å, α = 71.530(2)°, β = 84.594(2)°, γ = 72.106(2)°, U = 734.93(18) Å3, Z = 2, F(000) = 336, GOF = 1.042, R1 = 0.0396, wR2 = 0.0915 [I > 2σ(I)]. For 2: C10H15N9Zn, M = 326.70, triclinic, P, a = 7.2594(12) Å, b = 9.3403(16) Å, c = 11.835(2) Å, α = 71.216(2)°, β = 76.045(2)°, γ = 70.261(2)°, U = 707.2(2) Å3, Z = 2, F(000) = 336, GOF = 1.099, R1 = 0.0295, wR2 = 0.0709 [I > 2σ(I)]. For 3: C10H15N9Zn, M = 326.70, monoclinic, P21/c, a = 9.8594(17) Å, b = 11.0993(18) Å, c = 13.426(2) Å, α = 90°, β = 93.789(2)°, γ = 90°, U = 1466.0(4) Å3, Z = 4, F(000) = 672, GOF = 1.025, R1 = 0.0490, wR2 = 0.1268 [I > 2σ(I)]. , a = 8.3950(12) Å, b = 9.5035(13) Å, c = 10.2058(14) Å, α = 71.530(2)°, β = 84.594(2)°, γ = 72.106(2)°, U = 734.93(18) Å3, Z = 2, F(000) = 336, GOF = 1.042, R1 = 0.0396, wR2 = 0.0915 [I > 2σ(I)]. For 2: C10H15N9Zn, M = 326.70, triclinic, P, a = 7.2594(12) Å, b = 9.3403(16) Å, c = 11.835(2) Å, α = 71.216(2)°, β = 76.045(2)°, γ = 70.261(2)°, U = 707.2(2) Å3, Z = 2, F(000) = 336, GOF = 1.099, R1 = 0.0295, wR2 = 0.0709 [I > 2σ(I)]. For 3: C10H15N9Zn, M = 326.70, monoclinic, P21/c, a = 9.8594(17) Å, b = 11.0993(18) Å, c = 13.426(2) Å, α = 90°, β = 93.789(2)°, γ = 90°, U = 1466.0(4) Å3, Z = 4, F(000) = 672, GOF = 1.025, R1 = 0.0490, wR2 = 0.1268 [I > 2σ(I)]. |
| This journal is © The Royal Society of Chemistry 2011 |