Uranyl triazolate formation via an in situ Huisgen 1,3-dipolar cycloaddition reaction†
Karah E.
Knope
and
Christopher L.
Cahill
*
Department of Chemistry, The George Washington University, 725 21st, Street, NW, Washington, DC, USA. E-mail: cahill@gwu.edu; Fax: +1 (202) 994-5873; Tel: +1 (202) 994-6959
First published on 27th August 2010
Abstract
A two dimensional UO22+ coordination polymer, (UO2)3(C10H5N3O4)2(OH)2(H2O)2, has been synthesized under solvothermal conditions. The triazolate ligand, 1-(4-carboxyphenyl)-1H-1,2,3-triazole-4-carboxylic acid (CPTAZ) has been generated via a 1,3-dipolar cycloaddition of 4-azidobenzoic acid and propiolic acid. Reactions of the UO22+ cation with both the in situ generated triazolate ligand and the presynthesized ligand have been explored. The structure, fluorescent and thermal behaviour of this material are presented, as is a discussion of the utility of in situ ligand formation versus direct assembly.
Introduction
In situ ligand synthesis (ISLS) refers to a process wherein organic species undergo oxidation, reduction, hydrolysis or other reactions to yield a modified ligand that is subsequently observed in crystalline reaction products. The notion of generating ligands in situ during the synthesis of coordination polymers and as “a new approach to inorganic crystal engineering” was first proposed by Champness and Schroder et al. in 1997.1 Following their unexpected observation of the in situcyclisation of 1,2-trans-(4-pyridyl)ethene to form 1,2,3,4-tetrakis(4-pyridyl)cyclobutane, a variety of hybrid materials were prepared in this manner, many under hydro(solvo)thermal conditions.2 Others have sought to capitalize on this route since then by pointing out possible advantages over traditional syntheses (i.e. direct assembly wherein the organic ligands observed in the product are the same as those introduced as reactants), including simplified reaction schemes, one-pot syntheses, slow ligand generation to promote single crystal growth and even environmental friendliness. In situ ligand formation has even been shown in some cases to provide a pathway to materials that are not accessible through the direct reaction of the metal center with the organic linker.2–5 Most reports of ISLS are largely serendipitous, however, and little effort has been made to carefully explore the usefulness of this approach through controlled reactions. In other words, attempts to reproduce compounds obtained via ISLS using direct assembly are fairly limited.4,6–9 Moreover, many examples of in situ ligand formation are further complicated by convoluted speciation profiles resulting from the oxidation or decomposition of the organic ligand.10,11 The influence of these “spectator” species, generated concurrently with the ligand (yet not observed in crystalline products) on product formation is generally not considered despite many instances where such species have been shown to dramatically impact product formation.9,12–15One means of exploring in situ reactions without having to consider such decomposition products is to construct the ligand rather than generate it through a decomposition reaction.6,16,17 Click reactions are ideal candidates for this approach and have become an increasingly popular way to create organic molecules of diverse structure and function.18,19 In particular, the Huisgen 1,3-dipolar cycloaddition of alkynes to azides is well-suited to assemble ligands in situ.20Azides and alkynes are tolerant to a range of reaction conditions and can be easily functionalized without affecting reactivity. As such, they provide a relatively simple system for exploring in situ ligand formation in materials synthesis without the complication of decomposition products. Click reactions have been used successfully to prepare a variety of transition metal containing complexes yet direct assembly was not explored in these systems.15,21–25
In this work, we explored in situ ligand formation as a means of generating UO22+ containing hybrid materials. As the UO22+ cation is a relatively hard Lewis acid that tends to bind harder functional groups, carboxylate functionalized azides and alkynes were used as starting materials in this study. The in situ formation of the triazolate ligand 1-(4-carboxyphenyl)-1H-1,2,3-triazole-4-carboxylic acid (CPTAZ) is depicted in Scheme 1. The products obtained via the ISLS and direct assembly synthetic pathways have been compared in the context of assessing the utility of generating ligands in situ. The thermal and fluorescent properties of the resulting UO22+ triazolate have also been explored.
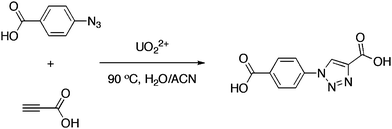 | ||
| Scheme 1 In situ triazole formation from 4-azidobenzoic acid and propiolic acid. | ||
Experimental
Synthesis
Caution: whereas the uranium oxyacetate (UO2)(CH3CO2)2·2H2O used in this investigation contains depleted U, standard precautions for handling radioactive substances should be followed. Compound 1, (UO2)3(OH)2(H2O)2(C10H5N3O4)2, was prepared through both direct assembly and in situ ligand formation as described below.Synthesis of CPTAZ ligand
The triazolate ligand, 1-(4-carboxyphenyl)-1H-1,2,3-triazole-4-carboxylic acid, was prepared from the reaction of 4-azidobenzoic acid (1.174 g, 7.2 mmol) and propiolic acid (0.372 mL, 6.0 mmol) in 40 mL of a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 H2O/THF (v/v) solution. The solution was stirred under refluxing conditions for 24 hours, after which the reaction was placed on ice. The precipitate was isolated by filtration to give CPTAZ as a white powder. The IR spectrum of the product (KBr) showed nearly complete disappearance of the azide peak at ∼2100 cm−1 and appearance of peaks at ca. 1518 and 1448 cm−1 suggestive of triazole formation. Elemental analysis (Galbraith Laboratories, Knoxville, TN) suggested that the product contained 75% CPTAZ and 25% unreacted 4-azidobenzoic acid, observed (calculated): C 51.21% (51.51%); N 19.60% (19.48%); H 3.00% (3.04%). The crude product was then used without further purification.
:50 H2O/THF (v/v) solution. The solution was stirred under refluxing conditions for 24 hours, after which the reaction was placed on ice. The precipitate was isolated by filtration to give CPTAZ as a white powder. The IR spectrum of the product (KBr) showed nearly complete disappearance of the azide peak at ∼2100 cm−1 and appearance of peaks at ca. 1518 and 1448 cm−1 suggestive of triazole formation. Elemental analysis (Galbraith Laboratories, Knoxville, TN) suggested that the product contained 75% CPTAZ and 25% unreacted 4-azidobenzoic acid, observed (calculated): C 51.21% (51.51%); N 19.60% (19.48%); H 3.00% (3.04%). The crude product was then used without further purification.
Synthesis of 1via direct assembly
Uranium oxyacetate dihydrate (0.148 g, 0.35 mmol), crude CPTAZ (0.080g, 0.35 mmol) and 4 mL of a 50/50 ACN:H2O (v/v) solution were placed into a 23 mL Teflon-lined Parr bomb; the initial pH of the solution was 2.5. The reaction vessel was then sealed and heated statically at 90 °C. After 5 days the reaction was removed from the oven and cooled to room temperature over 4 hours. Upon cooling to room temperature, a cloudy yellow liquid (pH 3.0) was decanted and yellow plate-like crystals were obtained. The product was washed with distilled water, ethanol and THF and then allowed to air dry at room temperature. Yield: 80% (based on uranium).
Synthesis of 1viain situ ligand formation
Compound 1, (UO2)3(OH)2(H2O)2(C10H5N3O4)2, was also prepared via the in situ formation of the triazolate ligand. Uranium oxyacetate dihydrate (0.149 g, 0.35 mmol), 4-azidobenzoic acid (0.119 g, 0.73 mmol), propiolic acid (0.038 mL, 0.61 mmol) and 4 mL of a 50/50 ACN:H2O (v/v) solution were placed into a 23 mL Teflon-lined Parr bomb; the initial pH of the solution was 2.3. The reaction vessel was then sealed and heated statically at 90 °C. After 5 days the reaction was removed from the oven and cooled to room temperature over 4 hours. Upon cooling to room temperature, a cloudy yellow liquid (pH 2.7) was decanted and yellow needles and a yellow powder were obtained. The product was washed with distilled water, ethanol and THF and then allowed to air dry at room temperature. Yield: 45% (based on uranium). Elemental analysis (Galbraith Laboratories, Knoxville, TN), observed (calculated): C 17.83% (17.89%); N 6.14% (6.26%); H 1.04% (1.20%). Main IR frequencies (KBr/cm−1): 3604, 3498, 3406, 3168, 2368 (w), 2130 (w), 1576, 1512, 1434, 1320, 1272, 1064, 928, 858, 780, 726.
X-Ray structure determination
A yellow plate-like crystal of 1 (0.10 mm × 0.04 mm × 0.04 mm) was isolated from the product obtained via ISLS and mounted on a MiTeGen micromount. Reflections were collected at 100 K on a Bruker SMART diffractometer equipped with an APEX II CCD detector using Mo Kα radiation (λ = 0.71073) and a combination of 0.5° ω and φ scans. The data were integrated and corrected for absorption using the APEX2 suite of crystallographic software.26 The compound was solved using direct methods and refined using SHELXL-9727 within the WinGX software suite.28 All non-hydrogen atoms were located using difference Fourier maps and were ultimately refined anisotropically. Hydrogen atoms residing on the carbon atoms of the –C6H4 and –C2HN3 rings of the CPTAZ ligand were placed in calculated positions and bond distances were fixed at 0.93 Å. Hydrogen atoms of the bound water molecule (O8) in 1 were located and refined with distance restraints of 0.82 Å. The hydrogen atom bound to O4 was also located in the difference Fourier map and refined with an O–H distance restraint of 0.80 Å.
Crystal data for compound 1. Mw = 1342.48, monoclinic, P21/n, a = 12.9037(11) Å, b = 6.1373(5) Å, c = 18.3478(16), β = 98.450(2)°, V = 1437.3(2) Å3, Z = 2, Dcalc = 3.102 Mg m−3, µ = 16.955 mm−1, 26513 reflections collected, 4090 independent [R(int) = 0.0456], Final R indices [I > 2σ(I)] R1 = 0.0215, wR2 = 0.0438, GooF = 1.029, largest diff. peak and hole 1.245 and −1.196 e− Å−3.†
Powder X-ray diffraction data were collected for the products obtained via both direct assembly and ISLS using a Rigaku Miniflex diffractometer (Cu Kα, 3–60°) and manipulated using the JADE software package.29 Agreement between the calculated and observed patterns (Fig. 1) suggests that the single crystal used for structure determination was representative of the bulk sample. Moreover the products synthesized by both direct assembly and in situ ligand formation reactions are the same.
 | ||
| Fig. 1 Powder X-ray diffraction spectra (shown from 5–32° 2θ, Cu Kα) for 1 synthesized via direct assembly (green) and in situ ligand formation (red). The calculated pattern is shown in black. | ||
Characterization
The emission spectrum for 1 was collected on a Shimadzu RF-5301 PC Spectrofluorophotometer (uranium excitation wavelength 365 nm; emission wavelength: 450–600 nm; slit width: 1.5 nm (excitation) and 1.5 nm (emission); sensitivity: high with a UV-35 filter). Thermogravimetric analysis (TGA) was performed on a Perkin Elmer Pyris 1 at a rate of 10 °C min−1 over a temperature range of 30–800 °C under flowing nitrogen gas. PXRD data, the fluorescence spectrum and the TGA plot can be found in the ESI†.Results
Structure description
Compound 1 is built from two unique U(VI) metal centers and one unique CPTAZ ligand as shown in Fig. 1. U1 is bound to two axial oxygen atoms, O1 and O2, at an average distance of 1.780Å to form the UO22+ moiety. Further, the UO22+ cation is equatorially coordinated to 4 oxygen atoms (O3, O4, O4iv, and O5) and one nitrogen atom (N1) to form an overall pentagonal bipyramidal coordination geometry. U2 alternatively adopts an overall hexagonal bipyramidal geometry with the uranyl oxygen atoms (O9 and O9ii) at an average distance of 1.779 Å. U2 is equatorially coordinated to six oxygen atoms, four (O6, O7 and their symmetry equivalents) from two bidentate CPTAZ units and two (O8 and its symmetry equivalent) from bound water molecules. Two U1 sites coordinate to hydroxyl oxygen atoms O4 and O4iv to form the edge-shared dimers (Fig. 2a) which are subsequently linked along [010] via the triazolate ligand. Additional coordination of the CPTAZ units to U2 results in 2D sheets (Fig. 3a). The sheets stack as shown in Fig. 3b. Selected bond lengths and angles are listed in Table 1.| a Superscript denotes symmetry transformations ii = x, y − 1, −z. | |||
|---|---|---|---|
| U1–O1 | 1.775(3) | U2–O7 | 2.436 |
| U1–O2 | 1.786(3) | U2–O8 | 2.493 |
| U1–O3 | 2.415(3) | U2–O6 | 2.467 |
| U1–O4 | 2.299(3) | ||
| U1–O5 | 2.373(3) | Bond angles | |
| U1–N1 | 2.605(3) | O1–U1–O2 | 174.37(12) |
| U2–O9 | 1.779(3) | O9–U2–O9ii | 180.00(11) |
 | ||
| Fig. 2 ORTEP illustration of 1. Ellipsoids are shown at 50% probability level. Hydrogen atoms have been omitted for clarity. Superscript denotes symmetry transformations i = −x + 5/2, y − 1/2, −z + 1/2; ii = x, y − 1, z; iii = −x + 1, −y, −z; iv = −x + 1, −y + 1, −z; v = x, y + 1, z; vi = −x + 3, −y + 1, −z + 1. | ||
![Polyhedral representation of 1 viewed down the (a) [1 0 −1] direction showing the topology of the 2-dimensional sheets and (b) [010] illustrating the stacking of the layers. Yellow polyhedra are U(vi) atoms in pentagonal and hexagonal bipyramidal geometry. Black, blue and red spheres represent the carbon, nitrogen, and oxygen atoms, respectively. Hydrogen atoms have been omitted for clarity.](/image/article/2011/CE/c0ce00231c/c0ce00231c-f3.gif) | ||
| Fig. 3 Polyhedral representation of 1 viewed down the (a) [1 0 −1] direction showing the topology of the 2-dimensional sheets and (b) [010] illustrating the stacking of the layers. Yellow polyhedra are U(VI) atoms in pentagonal and hexagonal bipyramidal geometry. Black, blue and red spheres represent the carbon, nitrogen, and oxygen atoms, respectively. Hydrogen atoms have been omitted for clarity. | ||
Powder X-ray diffraction
The powder patterns of the reaction products obtained via direct assembly and in situ ligand reactions are shown in Fig. 1. Comparison of the observed patterns reveals that the products assembled through the two synthetic routes are the same.Fluorescence studies
The emission spectrum for 1 showed weak uranyl fluorescence and exhibited characteristic vibronic structure of the UO22+ cation with peaks ranging from 475 to 600 nm. The emission spectrum for 1 is available in the ESI† as Fig. S3.Thermogravimetric analysis
The TGA curve for 1 exhibits four weight loss steps. The first step took place between 200 and 300 °C with an initial weight loss of ∼4%, consistent with the loss of the two bound water molecules and an additional molecule of H2O presumably from one of the –OH units. Decomposition of the triazolate ligand occurred in three steps beginning around ∼300 °C and complete by ∼800 °C. Loss of the triazolate ligand from the structure resulted in an additional weight loss of approximately 26%. The TGA curve for 1 is consistent with decomposition of the materials to multiple uranyl oxide phases that likely include UO3, UO2 and U3O7. Powder X-ray diffraction data of the resulting products support this finding. The TGA plot and powder diffraction data are available in the ESI† (Fig. S4 and S5).Discussion
Although ISLS has been a largely serendipitous process, there are many reported benefits of this synthetic route. For example, some products have been synthesized by ISLS that are not accessible via direct assembly.2,3 In some cases, this was attributed to the slow release of the ligand in situ, thereby promoting the formation of unique products. Alternatively, in situester hydrolysis has been explored in metal–phosphonate systems wherein the stability of metal–phosphate complexes has often made it difficult to obtain single crystals suitable for structure determination.4,12,13,30–32Most reports of in situ ligand formation including the ester hydrolysis mentioned above, however, have focused on hydrolysis, oxidation or decomposition reactions;2,3 systems in which the number of organic species in solution likely increases over time. To give another example, we previously reported the oxidation of DABCO as a means of preparing a UO22+–oxalate–glycolate.10 In this system, in situoxalate formation occurs via a complex reaction mechanism wherein decomposition or degradation of the organic species yields a potentially complicated organic speciation profile. Though efforts were made to elucidate the mechanism of product formation, we were unable to account for all of the species generated in situ. This was somewhat problematic considering the numerous examples wherein spectator species, charge balancing counter cations and templates have been found to influence product formation.9,12,14,33,34
In the click system, by contrast, the organic speciation profile is relatively simple. Click reactions and products assembled via these reactions rely on bond formation and as such the number of organic species in solution likely decreases over the course of the reaction. Thus, the number of “spectator” species is limited to the starting materials and the assembled click product. The fact that both in situ ligand formation and direct assembly synthetic routes yield the same product can perhaps be attributed to the absence of other, unaccounted for, organic species.
Differences between in situoxidation/hydrolysis/degradation reactions and click reactions may also be attributed to the availability of metal–ligand coordination sites. As mentioned previously, in situ ligand formation “slowly releases” the ligand over the course of the reaction. In other words, the building blocks needed for assembly and those that are ultimately observed in the final product are not present at the start of the reaction. The carboxylate functional groups are immediately available for coordination to the UO22+ cation in the click system, however, in systems wherein the ligand is generated through decomposition or degradation of the organic species, the starting organic often has no sites available for direct metal–ligand coordination. In these cases, the in situ generated ligands are “gently introduced” over the course of the reaction. Metal–ligand coordination is thus dependent on the rate of oxidation or hydrolysis and hence the availability of metal coordination sites. This would not necessarily be the case in click reactions where functional groups that are candidates for metal coordination are immediately available.
The primary benefit of generating the click product in situ, in this case, is that it provides an easy one-pot synthesis. Generating the triazolatein situ removes the need to presynthesize the ligand and also provides a facile route to a ligand that is not commercially available. More generally, click reactions also offer the ability to explore in situ ligand synthesis via controlled reactions. Admittedly this is not a tremendous advantage in this case but this observation highlights a significant difference between “construction” versus “destruction” reactions. That is, we must be cognizant of organic species present and evolving over the course of the reaction in order to fully understand factors that are contributing to phase formation and mechanisms of product formation. Further, these efforts are in fact control experiments that, with the exception of a few examples,5,9,10 have not been explored elsewhere.
Conclusion
In summary, we have prepared a novel 2-dimensional UO22+–triazolate wherein the CPTAZ unit was prepared via a 1,3-dipolar cycloaddition. The utility of in situ ligand synthesis as an alternative route for synthesizing hybrid materials has been examined through a controlled set of reactions. The product assembled viain situ ligand formation has been compared to the product synthesized by direct assembly and it was found that both synthetic approaches resulted in the same UO22+–triazolate product. Here the benefit of generating the triazolate product in situ is that it removes the need to presynthesize the organic ligand. We also note that this work provides only one data point and that inquiries into in situ click reactions are ripe for future exploration. Variables such as alkane chain length, rigidity, functionality and metal ion coordination modes can be surveyed within the click system by choosing appropriately functionalized azides and alkynes. Efforts in these areas are currently under investigation.Acknowledgements
This work was supported by (1) The Materials Science of Actinides, an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Office of Basic Energy Science under grant DE-SC0001089 and (2) The Chemical Sciences, Geosciences and Biosciences Division, Office of Science, Heavy Elements Program, US Department of Energy, under grant DE-FG02-05ER15736 at GWU. The X-ray diffraction instrumentation was purchased with the support from The National Science Foundation under grant DMR-0419754.References
- A. J. Blake, N. R. Champness, S. S. M. Chung, W.-S. Li and M. Schroder, Chem. Commun., 1997, 1675 RSC
.
- X.-M. Zhang, Coord. Chem. Rev., 2005, 249, 1201–1219 CrossRef CAS
- X.-M. Chen and M.-L. Tong, Acc. Chem. Res., 2006, 40, 162–170
- K. E. Knope and C. L. Cahill, Eur. J. Inorg. Chem., 2010, 8, 1177–1185 CrossRef
- Y.-T. Wang, H.-H. Fan, H.-Z. Wang and X.-M. Chen, Inorg. Chem., 2005, 44, 4148–4150 CrossRef CAS
- C. E. Rowland, N. Belai, K. E. Knope and C. L. Cahill, Cryst. Growth Des., 2010, 10, 1390–1398 CrossRef CAS
- K. L. Ziegelgruber, K. E. Knope, M. Frisch and C. L. Cahill, J. Solid State Chem., 2008, 181, 373–381 CrossRef CAS
- J. Y. Lu, J. Macias, J. Lu and J. E. Cmaidalka, Cryst. Growth Des., 2002, 2, 485–487 CrossRef CAS
- B. Li, W. Gu, L.-Z. Zhang, J. Qu, Z.-P. Ma, X. Liu and D.-Z. Liao, Inorg. Chem., 2006, 45, 10425–10427 CrossRef CAS
- K. E. Knope and C. L. Cahill, Inorg. Chem., 2007, 46, 6607–6612 CrossRef CAS
- X. Li, R. Cao, D. Sun, Q. Shi, W. Bi and M. Hong, Inorg. Chem. Commun., 2003, 6, 815–818 CrossRef CAS
- K. E. Knope and C. L. Cahill, Inorg. Chem., 2008, 47, 7660–7672 CrossRef CAS
- J.-J. Hou and X.-M. Zhang, Cryst. Growth Des., 2006, 6, 1445–1452 CrossRef CAS
- A.-G. D. Nelson, T. H. Bray, W. Zhan, R. G. Haire, T. S. Sayler and T. E. Albrecht-Schmitt, Inorg. Chem., 2008, 47, 4945–4951 CrossRef CAS
- J.-P. Zhang, Y.-Y. Lin, X.-C. Huang and X.-M. Chen, Dalton Trans., 2005, 3681–3685 RSC
- R. Murugavel, K. Baheti and G. Anantharaman, Inorg. Chem., 2001, 40, 6870–6878 CrossRef CAS
- F. Li, L. Xu, B. Bi, X. Liu and L. Fan, CrystEngComm, 2008, 10, 693–698 RSC
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS
- J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018–8021 CrossRef CAS
- J.-P. Zhang, Y.-Y. Lin, X.-C. Huang and X.-M. Chen, J. Am. Chem. Soc., 2005, 127, 5495–5506 CrossRef CAS
- L. Cheng, W.-X. Zhang, B.-H. Ye, J.-B. Lin and X.-M. Chen, Inorg. Chem., 2007, 46, 1135–1143 CrossRef CAS
- D.-W. Fu, W. Zhang and R.-G. Xiong, Cryst. Growth Des., 2008, 8, 3461–3464 CrossRef CAS
- X.-S. Wang, Y.-Z. Tang, X.-F. Huang, Z.-R. Qu, C.-M. Che, P. W. H. Chan and R.-G. Xiong, Inorg. Chem., 2005, 44, 5278–5285 CrossRef CAS
- L. Hsiu-Mei and C. Tsung-Yuan, Cryst. Growth Des., 2009, 9, 2988–2990 CrossRef CAS
- Apex2, Bruker-AXS, Madison, WI, 2008
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef
- JADE, V6.1, Materials Data Inc., Livermore, CA, 2001.
- G. B. Hix, B. M. Kariuki, S. Kitchin and M. Tremayne, Inorg. Chem., 2001, 40, 1477–1481 CrossRef CAS
- P.-A. Jaffrès, D. Villemin and V. Caignaert, Chem. Commun., 1999, 1997 RSC
- X.-M. Zhang, Eur. J. Inorg. Chem., 2004, 544–548 CrossRef CAS
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef CAS
- G. Férey, Chem. Mater., 2001, 13, 3084–3098 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Spectrum, fluorescence spectrum, TGA plot and PXRD data. CCDC reference numbers 778238. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0ce00231c |
| This journal is © The Royal Society of Chemistry 2011 |