DOI:
10.1039/C0CE00200C
(Paper)
CrystEngComm, 2011,
13, 215-222
Coordination polymers derived from a flexible bis(pyridylurea) ligand: conformational change of the ligand and structural diversity of the complexes†
Received
12th May 2010
, Accepted 8th July 2010
First published on 3rd September 2010
Abstract
The assembly of a bis(pyridylurea) ligand, N,N′-ethane-1,2-diylbis(3-pyridin-4-ylurea) (L), with Zn(AcO)2, CdCl2, CdSO4 or CuSO4 led to four coordination polymers, {[Zn(AcO)2L]·H2O·CH3OH}n (1), {[CdCl2L2]·2DMF}n (2), {[CdSO4L(H2O)3]·3H2O}n (3), and {[CuSO4L(H2O)2]·2H2O}n (4). Compound 1 is an infinite 1D zigzag chain with alternate Zn(AcO)2 units and L molecules. The cadmium(II) dichloro complex 2 features a corrugated sheet structure with a (4,4) net topology, while the sulfato complex 3 shows a unique 1O/2U interwoven 3D structure assembled from zigzag chains. The copper(II) complex 4 is an exceptional diamondoid network with an unusual 12-fold [6 + 6] interpenetration mode. Interestingly, the ligand shows the expected flexibility in the formation of the coordination polymers. In 1, 3 and 4, the central ethylene spacer adopts the anti conformations and is roughly linear, whereas in 2 it assumes a gauche form and exists as a V-shaped linker. The structural variation of these coordination polymers as well as the conformational change of the ligand in the presence of different counter anions and metal ions is discussed.
Introduction
Great interest has focused on coordination polymers not only because of their unique advantages in functional solid materials, ion exchange, catalysis and optics, etc., but also for their fascinating structures.1 The variety of self-assembled structures relies largely on the presence of suitable metal–ligand interactions and supramolecular contacts (hydrogen bonding and other weak interactions). By careful selection of metal ion ‘nodes’ and rigid organic ‘linkers’ with definite coordination preferences, numerous coordination networks with specific topologies have been successfully constructed.1b,2 On the other hand, the structures of metal complexes which contain relatively flexible bridging ligands are less predictable due to the possible supramolecular isomerism, such as the different conformations the ligand may adopt.2,3 Nevertheless, the flexible ligands could accommodate different coordination environments by adjusting their conformation, length or inherent angle of the terminal coordinative groups, thus leading to diverse structures of the metal–organic networks.4 The pyridyl moiety is probably the most popular building block for the construction of metal–organic networks due to its strong coordination ability to metal ions. Many oligo-pyridyl ligands with both rigid and flexible bridges have been used in the assembly of coordination architectures, and the structural topologies and chemical and physical properties of them were extensively studied.3
The oligo(pyridylurea) ligands, which contain two or more pyridyl ends separated by various linkers, have proven to be both promising anion receptors and excellent building blocks for novel supramolecular architectures.5 Recently, some bis(pyridylurea) ligands with different spacers between the two pyridylurea fragments have been used for the construction of metal–ligand supramolecular complexes.6 By varying the length of the alkyl spacer and the coordination site of the terminal pyridines (3- or 4-position), we have obtained various coordination networks, such as metallomacrocycles and helical chains,5d 3-fold interpenetrated MOFs5e and 1D → 1D interpenetrated coordination polymers.5f Herein, we report a bis(4-pyridylurea) receptor bearing a flexible ethylene spacer, N,N′-ethane-1,2-diylbis(3-pyridin-4-ylurea) (L) and four metal–organic coordination complexes constructed with L, including a 1D chain {[Zn(AcO)2L]·H2O·CH3OH}n (1), a 2D layered structure {[CdCl2L2]·2DMF}n (2), a 3D interwoven structure assembled from 1D zigzag chains, {[CdSO4L(H2O)3]·3H2O}n (3), and a diamondoid network with an unusual 12-fold [6 + 6] interpenetration mode, {[CuSO4L(H2O)2]·2H2O}n (4) (Scheme 1).
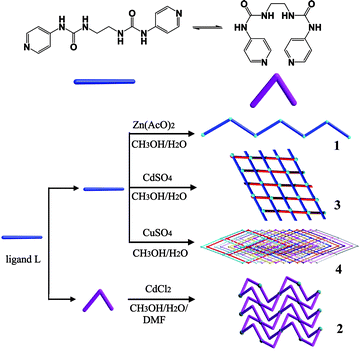 |
| | Scheme 1 Illustration of the conformation of L in compounds 1–4. | |
Results and discussion
The ligand N,N′-ethane-1,2-diylbis(3-pyridin-4-ylurea) (L) was synthesized from 4-isocyanatopyridine and 1,2-diaminoethane by a similar procedure to that reported previously for related ligands.5 Reaction of L with Zn(AcO)2·2H2O, CdCl2·2.5H2O, 3CdSO4·8H2O or CuSO4·5H2O afforded the complexes (1–4) with variable structures, which are dependent on the counter anion and metal ion employed. Very recently, the same ligand was reported, but it was synthesized by a different reaction of 4-aminopyridine and triphosgene in the presence of triethylamine.6f
Crystal structures
L·4H2O.
Colourless crystals (space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) of ligand L were obtained through slow evaporation of its water–methanol (1
) of ligand L were obtained through slow evaporation of its water–methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) solution. The asymmetric unit contains half ligand molecule and two crystal water molecules. The ligand adopts a roughly linear (GAG) conformation with torsion angles of 87.01°, −180.00° and −87.01° for the ethylene diamine spacer. Here the A (anti) or G (gauche) conformation is given when the N–C–C–N or C–C–N–C torsion angle (θ) is in the range 180 ≥ θ > 90° and 0 ≤ θ ≤ 90°, respectively (Fig. 1a).7 Interestingly, in the packed structure, each L molecule is surrounded by four infinite water chains through Nurea–H⋯Ow (N⋯O: 2.935(2)–2.950(2) Å), Ow–H⋯Npy (O⋯N: 2.766(2) Å) and Ow–H⋯OC
:1 v/v) solution. The asymmetric unit contains half ligand molecule and two crystal water molecules. The ligand adopts a roughly linear (GAG) conformation with torsion angles of 87.01°, −180.00° and −87.01° for the ethylene diamine spacer. Here the A (anti) or G (gauche) conformation is given when the N–C–C–N or C–C–N–C torsion angle (θ) is in the range 180 ≥ θ > 90° and 0 ≤ θ ≤ 90°, respectively (Fig. 1a).7 Interestingly, in the packed structure, each L molecule is surrounded by four infinite water chains through Nurea–H⋯Ow (N⋯O: 2.935(2)–2.950(2) Å), Ow–H⋯Npy (O⋯N: 2.766(2) Å) and Ow–H⋯OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (O⋯O: 2.762(3) Å) hydrogen bonds between the urea NH groups, the terminal pyridyl N atoms, and the urea carbonyl groups, respectively, and the water molecules (Fig. 1b). The ligands are further extended to an infinite chain bridged by two water molecules between two adjacent molecules (Fig. 1c), while the typical urea tape formed by self-association of the urea groups does not occur in this structure.
O (O⋯O: 2.762(3) Å) hydrogen bonds between the urea NH groups, the terminal pyridyl N atoms, and the urea carbonyl groups, respectively, and the water molecules (Fig. 1b). The ligands are further extended to an infinite chain bridged by two water molecules between two adjacent molecules (Fig. 1c), while the typical urea tape formed by self-association of the urea groups does not occur in this structure.
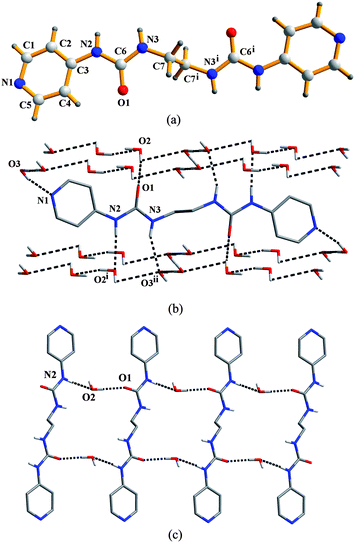 |
| | Fig. 1 (a) Molecular structure of L. Symmetry code: (i) 1 − x, 2 − y, 2 − z. (b) The four water chains around each L molecule. Hydrogen bond parameters (Å, °): O2⋯O1, 2.732(2); O2–H2A⋯O1, 172(3); O3⋯N1, 2.766(2); O3–H3A⋯N1, 166(3); N2⋯O2i, 2.935(2); N2–H2⋯O2i, 176.2; N3⋯O3ii, 2.950(2); N3–H3⋯ O3ii, 156.4. Symmetry codes: (i) 1 + x, y, z; (ii) 1 + x, 1 + y, 1 + z. (c) Part of the infinite chain of the ligand bridged by solvent water molecules. Hydrogen bond parameters (Å, °): N2⋯O2, 2.935(2); N2–H2⋯O2, 176.2; O2⋯O1, 2.732(2); O2–H2C⋯O1, 172(3). | |
{[Zn(AcO)2L]·H2O·CH3OH}n (1).
Slow evaporation of a solution of Zn(AcO)2·2H2O and L (Zn:L = 1:1) in CH3OH–H2O (v/v 1:1) afforded colourless block crystals of {[Zn(AcO)2L]·H2O·CH3OH}n (1). Complex 1 crystallizes in the triclinic space groupP. The asymmetric unit consists of one Zn atom, two AcO− ions, two halves of the ligand, one solvent CH3OH molecule and one water molecule. As shown in Fig. 2a, the Zn(II) ion is coordinated by two pyridyl nitrogen atoms from two ligands and four oxygen atoms of two chelating AcO− anions in a distorted octahedral geometry. The ligands act as bridges, linking the zinc atoms to form a one-dimensional zigzag chain (Fig. 2a) with a Zn⋯Zn separation of 19.62 Å and Zn⋯Zn⋯Zn angle of 114.68°. Like the free ligand, the L molecule adopts a roughly linear GAG conformation in 1. Selected bond lengths and bond angles for the complexes 1–4 are given in Table 1.
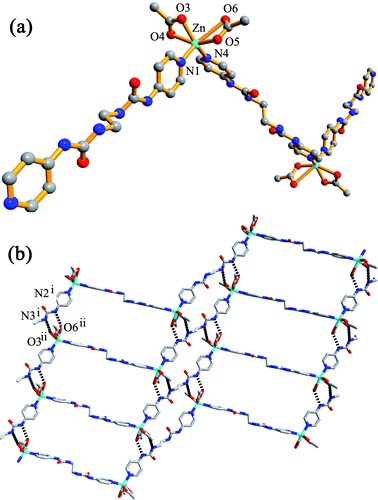 |
| | Fig. 2 (a) Part of the zigzag chain in 1; (b) 2D network formed by N–H⋯O hydrogen bonding. Hydrogen bond parameters (Å, °): N2i⋯O6ii, 2.8987(52); N2i–H2A⋯O6ii, 141.47; N3i⋯O3ii, 2.9521(61); N3i–H3⋯O3ii, 164.82. Symmetry codes: (i) −1 − x, 4 − y, 1 − z; (ii) −1 − x, 3 − y, 1 − z. | |
Table 1 Selected bond lengths (Å) and angles (°) for 1–4.a
|
Symmetry code: (A) x, −y, 0.5 + z.
|
| Compound 1 |
| Zn–N(1) |
2.049(3) |
Zn–O(3) |
2.066(3) |
| Zn–N(4) |
2.023(2) |
Zn–O(4) |
2.342(3) |
| Zn–O(5) |
2.016(2) |
O(5)–Zn–N(4) |
103.50(10) |
| O(5)–Zn–N(1) |
97.32(10) |
N(4)–Zn–N(1) |
108.48(11) |
| O(5)–Zn–O(3) |
96.94(11) |
N(4)–Zn–O(3) |
141.67(11) |
| N(1)–Zn–O(3) |
100.52(11) |
O(5)–Zn–O(4) |
153.13(11) |
| N(4)–Zn–O(4) |
94.36(10) |
N(1)–Zn–O(4) |
95.81(10) |
| O(3)–Zn–O(4) |
57.54(11) |
O(5)–Zn–O(6) |
51.079(78) |
| Compound 2 |
| Cd–N(1) |
2.3610(15) |
Cd–N(6)A |
2.3898(15) |
| Cd–Cl |
2.6294(6) |
N(1)–Cd–Cl |
89.91(4) |
| N(1)–Cd–N(6)A |
87.65(5) |
N(6)A–Cd–Cl |
89.67(4) |
| Compound 3 |
| Cd–O(6) |
2.285(3) |
Cd–N(4) |
2.312(3) |
| Cd–N(1) |
2.312(3) |
O(6)–Cd–N(4) |
93.81(12) |
| O(6)–Cd–N(1) |
89.54(11) |
N(4)–Cd–N(1) |
91.56(12) |
| Compound 4 |
| Cu–O(1) |
2.443(5) |
Cu–O(2) |
2.627(5) |
| Cu–O(3) |
1.991(3) |
Cu–O(5) |
1.975(3) |
| Cu–N(1) |
1.999(3) |
Cu–N(4) |
2.010(3) |
| O(5)–Cu–O(3) |
91.09(11) |
O(5)–Cu–N(1) |
175.77(13) |
| O(3)–Cu–N(1) |
89.76(12) |
O(5)–Cu–N(4) |
88.36(12) |
| O(3)–Cu–N(4) |
176.49(13) |
N(1)–Cu–N(4) |
90.54(13) |
In the extended structure, the zigzag chains are arranged parallel to each other and are further interlinked into a ladder-like two-dimensional network through N–H⋯O interactions between urea NH groups and oxygen atoms of the coordinated AcO− anions (Fig. 2b). Such hydrogen bonds play an important role in creating the higher-dimensional structure, as observed in many systems.8 A similar Zn(II) chloro complex with L, [(Zn(μ-L)Cl2)·X]n (X = disordered lattice solvents), was reported recently which is also a 1D zigzag polymeric chain.6f However, the ligand molecule adopts a G conformation with a disordered ethylene spacer in this complex, which is different from that (A) in 1. As a result, the zigzag shape of [(Zn(μ-L)Cl2)·X]n is determined by the ligands but not metal nodes, since the Zn atoms within each chain is co-linear rather than bent as in the present work.
{[CdCl2L2]·2DMF}n (2).
The reaction of L and CdCl2 in CH3OH–H2O–DMF (v/v/v 1:1:1) gave the complex {[CdCl2L2]·2DMF}n (2) as colourless block crystals crystallizing in the monoclinic space groupC2/c. The asymmetric unit contains half a Cd atom, one Cl atom, one ligand and one solvent DMF molecule. In the crystal structure, the Cd(II) center resides on a center of inversion, being coordinated by four pyridyl nitrogen atoms of four ligands and two chloride ions in an octahedral geometry (Fig. 3a). Unlike that in complex 1, the ligand adopts a V-shaped AGA conformation in 2.
 |
| | Fig. 3 Crystal structure of 2. (a) The coordination environment of the cadmium atom; (b, c) top view and side view of the corrugated sheet with the (4,4) net topology; (d) Hydrogen bonds between the 2D sheets; (e) Topological representation of the (4,4) connected network. Symmetry codes: (i) 0.5 − x, 0.5 − y,1 − z; (ii) x, −y, 0.5 + z; (iii) 0.5 − x, 0.5 + y, 0.5 − z. | |
The structure of this complex can be best described as a corrugated 2D sheet in which four ligands bridge four Cd(II) centers to form a (4,4) network topology (Fig. 3b). It is noteworthy that the (4,4) nets are one of the most frequently occurring motifs in metal coordination polymers; however most of them are constructed with rigid organic linkers, which result in largely planar two-dimensional grids. In contrast, the structures of corrugated grid-like sheet are not very common.1d,9 In the current case, the flexible ligand with the bent conformation it adopts is responsible for the corrugated (4,4) grids which show an approximate chair conformation (one unit is shown in yellow in Fig. 3b). Moreover, these corrugated sheets are further linked by N–H⋯O (N4⋯O1 = 2.862 Å) hydrogen bonds of the urea groups from adjacent layers, and the solvent DMF molecules are trapped between the layers by N–H⋯O hydrogen bonds with the urea groups (Fig. 3d).
{[CdSO4L(H2O)3]·3H2O}n (3).
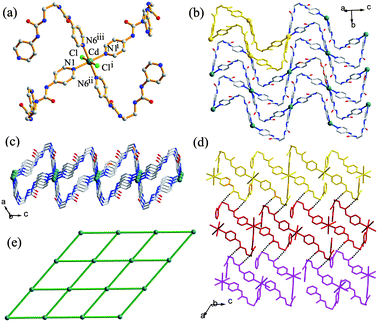
When the counter anion was changed from Cl− to SO42−, the complex {[CdSO4L(H2O)3]·3H2O}n (3) was obtained as colourless block crystals through diffusion of a solution of L in CH3OH and a solution of CdSO4 in H2O. Complex 3 crystallizes in the monoclinic space groupC2/c, with one Cd atom, one SO42− ion, two halves of the ligand and six water molecules (three coordinated and three crystallized) in an asymmetric unit. Compared to the cadmium chloro complex 2, in which the Cd2+ ion is coordinated by four ligands and two anions, the octahedral Cd2+ center in 3 is coordinated by only two L ligands together with a monodentate SO42− anion and three H2O molecules (Fig. 4a). The ligand molecules show a linear AAA conformation, which is different from that in 1 and 2. The two ligands that coordinate to the metal center in a cis fashion further bridge other Cd atoms to form a one-dimensional zigzag chain structure (Fig. 4b) rather than the 2D (4,4) net in 2. The Cd⋯Cd separation is 20.21 Å and Cd⋯Cd⋯Cd angle is 70.53° in each chain.
 |
| | Fig. 4 (a) The molecular structure of 3 showing the local coordination geometry around the metal center. (b) Perspective view of the 1D coordination polymeric strand. (c) 1O/2U interwoven 2D network. (d) Top view of the schematic representation of the 1O/2U cloth-like sheet. (e) Schematic representation of the 3D network in 3. | |
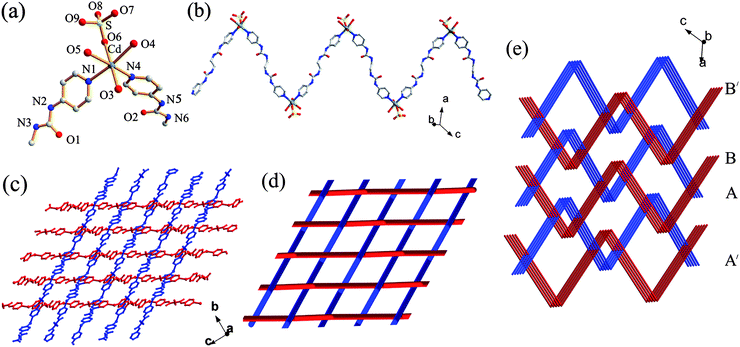
An unusual feature of this compound is that the zigzag chains interweave in a ‘one-over/two-under’ (1O/2U) fashion to create a cloth-like sheet structure as shown in Fig. 4c and 4d. In such a supramolecular entanglement, each chain is chemically independent but physically interwoven with the perpendicular chains.10 Thus, in contrast to the interpenetrated networks, breaking of chemical bonds is not required to take apart this network structure. It is noteworthy that only a few interwoven “warp-and-woof” sheet networks of zigzag chains have been reported so far.11 For example, Ciani and co-workers11a described a 2D sheet (1O/1U) which represents the first interwoven network of 1D rigid zigzag coordination strands. Li et al.11b reported a 2D interwoven network (2O/2U), while Cheng et al.11c illustrated how the 1D zigzag chains in four different directions were hierarchically entangled to generate an unprecedented 3D interwoven framework. Very recently, Peedikakkal et al.11d reported a 1O/1U interwoven fabric structure formed by spiral 1D coordination polymeric chains of the PbII complex. To the best of our knowledge, compound 3 is the first 1O/2U interwoven fashion in coordination networks. In complex 3, every interwoven sheet containing chains propagating along two directions (A and B in Fig. 4e) further interweaves with the chains (B′ and A′) in the neighbouring sheets, also in the 1O/2U mode, and an overall 3D interlocking network is formed (Fig. 4e).
{[CuSO4L(H2O)2]·2H2O}n (4).
Needle-like blue crystals of the copper(II) complex 4 were obtained through slow diffusion of CuSO4·5H2O and L in CH3OH–H2O. Compound 4 crystallizes in the orthorhombic space groupPnna. The asymmetric unit consists of one Cu atom, two halves of SO42− ion, two halves of the ligand, and four water molecules (two coordinated and two crystallized) with the composition {[CuSO4L(H2O)2]·2H2O}n. Notably, complex 4 features a 12-fold [6 + 6] interpenetrated diamondoid net. According to the classification of the interpenetration patterns proposed by Blatov et al., the structure belongs to the interpenetration class IIIa. For class III (translational and non-translational), the overall entanglement is generated both by pure translations (TIVs, translating interpenetration vector; or PIVs, partial interpenetration vector) and by space group symmetry elements (NISEs, non-translating interpenetration symmetry element; or PISEs, partial interpenetration symmetry element). If a TIV exists, the array belongs to class IIIa.12
The diamond structure, as one of the most common and important types of topology, has attracted great attention for its potential applications in non-linear optics (NLO) since the pioneering work of Ermer.13 For the diamondoid coordination polymers, the n-fold interpenetrated networks have been an invariable highlight. As reviewed by Robson,14 the common interpenetration mode of diamond structure is that the nodes of independent nets are aligned and equally spaced along one of the 2-fold axes. However, exceptional interpenetration modes have also been reported in the literature.13b,15 Recently, Hsu et al.15a reported a 12-fold interpenetrated diamondoid net with a flexible ligand N,N′-di(4-pyridyl)adipoamide, [CuSO4(L′)(H2O)2]∞, which is the maximum number of interpenetration presently known for coordination polymers, though a hydrogen-bonded structure has been reported very recently to show a 18-fold interpenetration of diamond frame.16
Compound 4 represents the second example of the highest interpenetrating number (12) for diamondoid coordination polymers. Although compound 4 shows a similar 3D diamond-like topology as the reported first 12-fold interpenetrated diamondoid compound [CuSO4(L′)(H2O)2]∞, its composition and the local coordination environment of the Cu2+ ions are significantly different from the latter.15a Compound 4 contains two coordinated and two crystal waters molecules, respectively, while the reported first example has only one coordinated and one crystal water molecule. Consequently, the Cu2+ ions in 4 have a six-coordinate, Jahn–Teller distorted octahedral geometry, being coordinated by two nitrogen atoms from two bridging ligands, two oxygen atoms from two bridging sulfate ions, and two water molecules on the axial positions (Fig. 5a). In contrast, the Cu2+ centers in [CuSO4(L′)(H2O)2]∞ show a five-coordinate distorted square pyramidal geometry with only one coordinated water (in the apical position).15a The Cu–Ow distances (Cu–O1, 2.443(5) Å; Cu–O2, 2.627(5) Å) in 4, however, are longer than that in the latter (2.275(5) Å). Like the case in its analogue [CuSO4(L′)(H2O)2]∞, the SO42− anions in 4 also adopt the μ2-η1,η1-bridging coordination fashion, linking two Cu ions to form an eight-membered cycle with a comparable Cu⋯Cu separation (4.797 Å compared to 4.681 Å in [CuSO4(L′)(H2O)2]∞) (Fig. 5a). When the midpoint of the two SO4-bridged copper atoms is viewed as a connecting node, we can see that it acts as a tetrahedral node which connects four other neighbours through four bis(pyridylurea) ligands (Fig. 5b). The ligand molecules show two different conformations (AAA and GAG) in 4, and the adjoining nodes are separated by two different distances of 24.03 and 23.46 Å, respectively, which are similar to the reported compound (24.46 and 23.62 Å). The structure of 4 consists of a three-dimensional framework with the extended diamondoid topology. The maximum dimensions of the adamantane cage (37.72 × 80.92 × 32.28 Å3) are similar to the reported compound (37.36 × 82.27 × 32.61 Å3).15a Such a large cavity allows the unusual 12-fold interpenetration of the networks. This interpenetration mode differs from the normal mode and can be described as two sets of normal 6-fold nets. Fig. 5c shows a view from the b axis, in which the two sets of the normal sixfold net are interpenetrated with a relative translation vector of a/2 + b/2.2a,15a Thus, this mode is named a [6 + 6] interpenetrated diamondoid system.
![Crystal structure of 4. (a) The coordination environment around the Cu(ii) ions. Symmetry codes: (i) x, 1.5 − y, 1.5 − z; (ii) −x, 1 − y, −z; (iii) 1 − x, 2 − y; −z; (b) Schematic views of the 12-fold interpenetration; (c) The [6 + 6] interpenetration viewed from the b axis; (d) Hydrogen bonds around the sulfate ion. Hydrogen bond parameters (Å, °): N2i⋯O6, 2.853(4); N2i–H2Ai⋯O6, 165.5; O1⋯O6, 2.966(6); O1–H1Bi⋯O6, 145(5); N3i⋯O9, 2.864(6); N3i–H3i⋯O9, 148.9; O9⋯O5i, 2.795(5); O9–H9C⋯O5i, 127(5). Symmetry code: (i) x, 1.5 − y, 1.5 − z.](/image/article/2011/CE/c0ce00200c/c0ce00200c-f5.gif) |
| | Fig. 5 Crystal structure of 4. (a) The coordination environment around the Cu(II) ions. Symmetry codes: (i) x, 1.5 − y, 1.5 − z; (ii) −x, 1 − y, −z; (iii) 1 − x, 2 − y; −z; (b) Schematic views of the 12-fold interpenetration; (c) The [6 + 6] interpenetration viewed from the b axis; (d) Hydrogen bonds around the sulfate ion. Hydrogen bond parameters (Å, °): N2i⋯O6, 2.853(4); N2i–H2Ai⋯O6, 165.5; O1⋯O6, 2.966(6); O1–H1Bi⋯O6, 145(5); N3i⋯O9, 2.864(6); N3i–H3i⋯O9, 148.9; O9⋯O5i, 2.795(5); O9–H9C⋯O5i, 127(5). Symmetry code: (i) x, 1.5 − y, 1.5 − z. | |
Noticeably, the framework is supported by extensive hydrogen bonds. Each μ2-coordinated sulfate ion is surrounded by two urea fragments and four water molecules (both coordinated and solvent), accepting six hydrogen bonds, including two direct N–H⋯O contacts (N⋯O = 2.853, 2.921 Å) with the urea NH groups, two O–H⋯O bonds (O⋯O = 2.866, 2.966 Å) with the crystal water molecules, and two O–H⋯O contacts (O⋯O = 2.794, 2.916 Å) with the coordinated water molecules, respectively (Fig. 5d). Notably, the two urea groups do not display the eight-membered R22(8) hydrogen bonding mode, but donate only a single N–H⋯O hydrogen bond using one of the NH donors, while the other NH is bridged to the sulfate ion through an O–H⋯O interaction with a crystal water molecule. In addition, different O–H⋯O interactions from the urea carbonyls of the ligand L (O⋯O = 2.866 Å) or the crystal water (O⋯O = 2.806 Å) to the coordinated water molecules are also observed. It is noticed that there are only four hydrogen bonds around the sulfate ion in the first 12-fold diamondoid network.15a The enhanced anion binding in our compound 4 should be the result of the existence of two more water molecules (one coordinated and one crystal), which can serve as good hydrogen bond donors and acceptors.
PXRD and TG analyses of the complexes
Compounds 1, 2, 3 and 4 were further confirmed by elemental analyses, IR spectroscopy and powder X-ray diffraction. The PXRD patterns (Fig. 6) revealed the phase purity of the complexes. The thermal stability of the compounds was studied by TGA (Fig. 7). Compound 1 showed a slight weight loss from room temperature to 119 °C corresponding to the release of a CH3OH and a H2O molecule (observed weight loss 9.4%, calculated 10.8%), as well as a major weight loss occurring at above 208 °C due to the decomposition of the organic ligand. Compound 2 began to decompose from about 210 °C due to release of the two DMF molecules and loss and/or combustion of the organic components. The TGA curve of 3 showed a weight loss from room temperature to 151 °C corresponding to five water molecules (observed 14.0%, calculated 15.0%). The compound decomposed at 230 °C. Complex 4 has a reasonable stability in the solid state. There is a 13.0% weight loss in the temperature range 49–195 °C, which is attributed to the loss of the two coordinated and two crystal water molecules (calculated 13.5%). The compound began to decompose at above 241 °C with a sharp weight loss. It should be noted that the decomposition temperature of the first 12-fold diamondoid network is relatively low (around 80 °C).15a Thus, complex 4 exhibits an improved stability, which is probably due to the enhanced hydrogen bonding interactions in 4 as mentioned above.
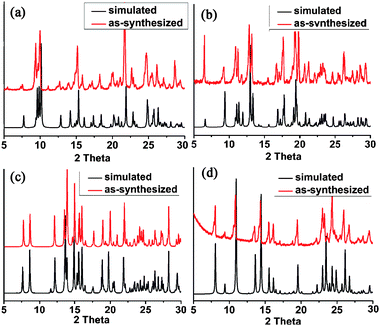 |
| | Fig. 6
Powder X-ray diffraction patterns: as-synthesized (red) and simulated from the single-crystal diffraction data (black): (a) 1; (b) 2; (c) 3; (d) 4. | |
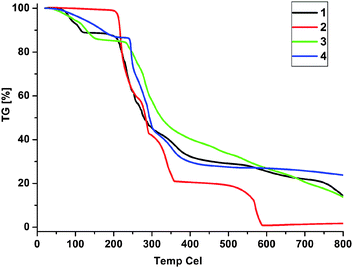 |
| | Fig. 7
TGA curves of the compounds 1–4. | |
Conclusions
We report four coordination polymers constructed from a flexible bis(pyridylurea) ligand (L). The supramolecular structures of the complexes vary from 1D zigzag chain (1) to 2D corrugated sheet (2) and interwoven 3D network (3), and then to a rare 3D diamondoid network with a 12-fold [6 + 6] interpenetration mode (4). Interestingly, the flexible ligand adopts different conformations in the complexes (roughly linear in 1, 3, and 4, and V-shaped in 2), and geometric needs of the metal atoms and coordination of the anions also play important roles in the construction of the complexes. Moreover, the urea groups participate in various hydrogen bonding interactions in all the complexes, which are essential in the formation of the secondary (or higher-dimensional) supramolecular structures.
Experimental
General procedures
All starting chemicals were commercially available and were used as received. Elemental analyses were done on a VarioEL instrument from Elementaranalysensysteme GmbH. IR spectra were recorded on a Bruker IFS 120HR spectrometer as KBr disks (500–4000 cm−1). 1H NMR spectra were recorded at ambient temperature on a Mercury Plus 400 MHz FT spectrometer in DMSO-d6. Melting points were detected on an X-4 Digital Vision MP Instrument. Powder X-ray diffraction (PXRD) data were collected on a Philips X'Pert PRO SUPER diffractometer with Cu-Kα radiation (λ = 1.54187 Å). TG analyses were carried out with a Pyris diamond instrument (Perkin Elmer) under N2 atmosphere with a heating rate of 10 °C min−1.
Synthesis of L.
4-Pyridyl acyl azide (3.00 g, 20.2 mmol) in freshly dried toluene (100 mL) was refluxed under a nitrogen atmosphere for 1 h to give a pale yellow solution. Then 1,2-diaminoethane (0.61 g, 10.1 mmol) was added and the mixture was refluxed for 30 min and then cooled to r.t. A white powder was collected and washed with toluene, diethyl ether and dried in vacuo. Yield: 1.5 g (50%). M.p.: 245–247 °C. Anal. calcd for C14H16N6O2·0.7H2O: C 53.73, H 5.60, N 26.80. Found: C 53.41, H 5.25, N 26.98%. 1H NMR (DMSO-d6, 400 MHz): 9.05 (s, 2 H, pyridyl-NH-), 8.28 (d, 4 H, J = 6.4 Hz, Py-H2), 7.37 (dd, 4 H, J = 1.4, 6.4 Hz, Py-H3), 6.46 (s, 2 H, CH2–NH-), 3.21 (t, 4 H, J = 2.8 Hz, urea-CH2). 13C NMR (DMSO-d6, 100 MHz): 154.8 (CO), 149.9 (Py-C4), 147.0 (Py-C2), 111.8 (Py-C3), 39.3 (urea-CH2-). IR (KBr pellet, cm−1): 3312 (N–H), 3087, 1681 (CO), 1601, 1534, 1211, 1004, 826, 530.
Synthesis of {[Zn(AcO)2L]·H2O·CH3OH}n (1).
The ligand L (20 mg, 0.07 mmol) and Zn(OAc)2·2H2O (12.2 mg, 0.07 mmol) were dissolved in a mixed solvent of CH3OH–H2O (1:1 v/v, 5 mL). Slow evaporation at r.t. for several days yielded colourless crystals of 1 (27 mg, 71%). M.p.: 206–207 °C. Anal. calcd for Zn(AcO)2L·H2O·CH3OH: C 42.75, H 5.29, N 15.74%. Found: C 42.42, H 4.94, N 15.93%. IR (KBr pellet, cm−1): 3357 (N–H), 3157, 3010, 2946, 1714 (CO), 1600, 1507, 1437, 1270, 1201, 1026, 835, 682, 632, 532.
Synthesis of {[CdCl2L2]·2DMF}n (2).
The ligand L (20 mg, 0.07 mmol) and CdCl2·2.5H2O (15.2 mg, 0.07 mmol) were dissolved in methanol–water–DMF (1:1:1 v/v/v, 5 mL). Slow evaporation at r.t. for a month yielded colourless crystals of 2 (14 mg, 42%). M.p.: > 300 °C. Anal. calcd for CdCl2L2·2DMF: C 43.90, H 4.98, N 21.08%. C 43.85, H 4.89, N 20.78%. IR (KBr pellet, cm−1): 3367 (N–H), 3068, 1699 (CO), 1595, 1517, 1427, 1263, 1208, 1096, 1011, 835, 715, 537.
Synthesis of {[CdSO4L(H2O)3]·3H2O}n (3).
A methanol solution (5 mL) of L (10 mg, 0.03 mmol) was layered on top of an aqueous solution of 3CdSO4·8H2O (8.5 mg, 0.01 mmol), and the mixture was allowed to diffuse at room temperature. After two weeks, colourless needle-like crystals were obtained at the interface. Yield: 8.5 mg (46%). M.p.: > 300 °C. Anal. calcd for CdSO4L·5H2O: C 28.08, H 4.38, N 14.03%. Found: C 28.47, H 4.53, N 14.00%. IR (KBr pellet, cm−1): 3328 (N–H), 3086, 1696 (CO), 1599, 1527, 1435, 1338, 1264, 1213, 1092, 1016, 829, 602, 529.
Synthesis of {[CuSO4L(H2O)2]·2H2O}n (4).
A methanol solution (5 mL) of L (30 mg, 0.1 mmol) was layered on the top of an aqueous solution (5 mL) of CuSO4·5H2O (25 mg, 0.1 mmol), and the mixture was allowed to diffuse at room temperature. Blue block crystals were formed at the interface in several days. Yield: 26 mg, (50%). Mp: 230–231 °C. Anal. calcd for CuSO4L·3H2O: C 32.72, H 4.31, N 16.35. Found: C 32.56, H 4.42, N 16.17%. FT-IR (KBr pellet, cm−1): 3399 (N–H), 3253, 1688 (CO), 1601, 1519, 1209, 1092, 1027, 842, 533.
Data collection was performed on a Bruker-AXS SMART CCD area detector diffractometer at 293 K using ω rotation scans with a scan width of 0.3° and Mo-Kα radiation (λ = 0.71073 Å). Multi-scan corrections were applied using SADABS.17 Structure solutions and refinements were performed with the SHELX-97 package.18 All non-hydrogen atoms were refined anisotropically by full-matrix least-squares on F2. The hydrogen atoms were included in idealized geometric positions with thermal parameters equivalent to 1.2 times those of carbon and nitrogen atoms. Hydrogen atoms of water molecules in L and 1 were located from the difference Fourier map and then refined by restraints (O–H = 0.85(1) Å), with U(H) fixed at 0.08 Å2. Crystallographic data for the compounds are summarized in Table 2.
| Compound |
L |
1
|
2
|
3
|
4
|
| Empirical formula |
C14H24N6O6 |
C19H28N6O8Zn
|
C34H46CdCl2N14O6 |
C14H28CdN6O12S |
C14H24CuN6O10S |
|
FW /g mol−1 |
372.39 |
533.84 |
930.15 |
616.88 |
531.99 |
| Crystal system |
Triclinic |
Triclinic |
Monoclinic |
Monoclinic |
Orthorhombic |
| Space group |
P
|
P
|
C2/c |
C2/c |
Pnna
|
|
a /Å |
7.2549(13) |
10.447(2) |
31.510(7) |
30.671(12) |
18.861(5) |
|
b /Å |
7.7629(15) |
10.755(3) |
8.534(2) |
7.551(3) |
16.141(4) |
|
c /Å |
9.1054(16) |
12.473(3) |
18.891(4) |
24.101(10) |
13.487(4) |
|
α /° |
98.095(3) |
104.330(4) |
90.00 |
90.00 |
90.00 |
|
β /° |
99.513(3) |
102.140(4) |
122.100(2) |
122.271(4) |
90.00 |
|
γ /° |
110.281(3) |
110.924(3) |
90.00 |
90.00 |
90.00 |
|
V /Å3 |
463.39(15) |
1197.3(5) |
4303.4(17) |
4720(3) |
4106.0(18) |
|
Z
|
1 |
2 |
4 |
8 |
8 |
|
D
c /g cm−3 |
1.334 |
1.481 |
1.436 |
1.736 |
1.721 |
| Crystal size /mm |
0.30 × 0.25 × 0.22 |
0.30 × 0.28 × 0.25 |
0.40 × 0.35 × 0.30 |
0.25 × 0.20 × 0.20 |
0.40 × 0.25 × 0.20 |
|
F(000) |
198 |
556 |
1912 |
2512 |
2200 |
|
μ /mm−1 |
0.11 |
1.08 |
0.69 |
1.09 |
1.23 |
|
θ range |
2.32–25.00 |
1.79–25.97 |
1.53–28.20 |
1.57–25.55 |
1.86–26.62 |
| Reflections collected |
2373 |
6647 |
17308 |
15660 |
22679 |
| Independent reflections |
1620 |
4614 |
5184 |
4383 |
4299 |
| Observed reflections [I > 2σ(I)] |
1410 |
3353 |
4634 |
3125 |
2552 |
|
R
int
|
0.018 |
0.027 |
0.018 |
0.057 |
0.084 |
|
R
1; wR2 [I > 2σ(I)] |
0.0477, 0.1654 |
0.0462, 0.0941 |
0.0269, 0.0693 |
0.0396, 0.0787 |
0.0486, 0.1190 |
|
R
1; wR2 (all data) |
0.0535, 0.1729 |
0.0722, 0.1042 |
0.0313, 0.0721 |
0.0665, 0.0888 |
0.0986, 0.1450 |
| GOF (F2) |
1.384 |
1.015 |
1.062 |
1.024 |
1.045 |
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 20872149).
References
-
(a) L. Carlucci, G. Ciani and D. M. Proserpio, Coord. Chem. Rev., 2003, 246, 247–289 CrossRef CAS;
(b) N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176–182 CrossRef CAS;
(c) B.-H. Ye, M.-L. Tong and X.-M. Chen, Coord. Chem. Rev., 2005, 249, 545–565 CrossRef CAS;
(d) A. Y. Robin and K. M. Fromm, Coord. Chem. Rev., 2006, 250, 2127–2157 CrossRef CAS;
(e) C. Janiak, Dalton Trans., 2003, 2781–2804 RSC.
-
(a) C.-W. Yeh, J.-D. Chen and J.-C. Wang, Polyhedron, 2008, 27, 3611–3618 CrossRef CAS;
(b) K. Biradha, M. Sarkar and L. Rajput, Chem. Commun., 2006, 4169–4179 RSC.
-
(a) L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, Chem. Commun., 2000, 1319–1320 RSC;
(b) Z. Qin, M. C. Jennings and R. J. Puddephatt, Chem.–Eur. J., 2002, 8, 735–738 CrossRef CAS;
(c) Z. R. Ranjbar and A. Morsali, J. Mol. Struct., 2009, 936, 206–212 CrossRef CAS;
(d) F. Bigdeli, A. Morsali and P. Retailleau, Polyhedron, 2010, 29, 801–806 CrossRef CAS;
(e) J.-D. Lin, Z.-H. Li, J.-R. Li and S.-W. Du, Polyhedron, 2007, 26, 107–114 CrossRef CAS;
(f) H.-Z. Dong, J. Zhao, S.-H. Gou and H.-B. Zhu, Polyhedron, 2009, 28, 1040–1048 CrossRef CAS;
(g) Y. Gong, R. Wang, D. Yuan, W. Su, Y. Huang, C. Yue, F. Jiang and M. Hong, Polyhedron, 2007, 26, 5309–5316 CrossRef CAS;
(h) J. Wang, M. Huang, P. Liu and W. Cheng, J. Mol. Struct., 2008, 875, 22–26 CrossRef CAS;
(i) Y.-F. Hsu, H.-L. Hu, C.-J. Wu, C.-W. Yeh, D. M. Proserpio and J.-D. Chen, CrystEngComm, 2009, 11, 168–176 RSC.
-
(a) J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670–4679 CrossRef CAS;
(b) D. B. Cordes and L. R. Hanton, Inorg. Chem., 2007, 46, 1634–1644 CrossRef CAS.
-
(a) B. Wu, X. Huang, Y. Xia, X.-J. Yang and C. Janiak, CrystEngComm, 2007, 9, 676–685 RSC;
(b) B. Wu, J. Liang, J. Yang, C. Jia, X.-J. Yang, H. Zhang, N. Tang and C. Janiak, Chem. Commun., 2008, 1762–1764 RSC;
(c) Y. Xia, B. Wu, Y. Liu, Z. Yang, X. Huang, L. He and X.-J. Yang, CrystEngComm, 2009, 11, 1849–1856 RSC;
(d) J. Liang, B. Wu, C. Jia and X.-J. Yang, CrystEngComm, 2009, 11, 975–977 RSC;
(e) B. Wu, J. Liang, Y. Zhao, M. Li, S. Li, Y. Liu, Y. Zhang and X.-J. Yang, CrystEngComm, 2010, 12, 2129–2134 RSC;
(f) S. Li, B. Wu, Y. Hao, Y. Liu and X.-J. Yang, CrystEngComm, 2010, 12, 2001–2004 RSC.
-
(a) J. M. Russell, A. D. M. Parker, I. Radosavljevic-Evans, J. A. K. Howard and J. W. Steed, Chem. Commun., 2006, 269–271 RSC;
(b) P. Byrne, G. O. Lloyd, N. Clarke and J. W. Steed, Angew. Chem., Int. Ed., 2008, 47, 5761–5764 CrossRef CAS;
(c) L. Applegarth, A. E. Goeta and J. W. Steed, Chem. Commun., 2005, 2405–2406 RSC;
(d) R. Custelcean, V. Sellin and B. A. Moyer, Chem. Commun., 2007, 1541–1543 RSC;
(e) P. Byrne, D. R. Turner, G. O. Lloyd, N. Clarke and J. W. Steed, Cryst. Growth Des., 2008, 8, 3335–3344 CrossRef CAS;
(f) N. N. Adarsh, D. K. Kumar and P. Dastidar, Cryst. Growth Des., 2009, 9, 2979–2983 CrossRef CAS.
-
(a) L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, CrystEngComm, 2002, 4, 121–129 RSC;
(b) H.-C. Chen, H.-L. Hu, Z.-K. Chan, C.-W. Yeh, H.-W. Jia, C.-P. Wu, J.-D. Chen and J.-C. Wang, Cryst. Growth Des., 2007, 7, 698–704 CrossRef CAS.
-
(a) D. Min, B.-Y. Cho and S. W. Lee, Inorg. Chim. Acta, 2006, 359, 577–584 CrossRef CAS;
(b) H. Zhu, C. Huang, W. Huang and S. Gou, Inorg. Chem. Commun., 2004, 7, 1095–1099 CrossRef CAS.
-
(a) Y.-H. Wang, K.-L. Chu, H.-C. Chen, C.-W. Yeh, Z.-K. Chan, M.-C. Suen, J.-D. Chen and J.-C. Wang, CrystEngComm, 2006, 8, 84–93 RSC;
(b) M. A. Braverman and R. L. LaDuca, Cryst. Growth Des., 2007, 7, 2343–2351 CrossRef CAS;
(c) F. Dai, H. He, D. Gao, F. Ye, X. Qiu and D. Sun, CrystEngComm, 2009, 11, 2516–2522 RSC;
(d) G.-P. Yang, Y.-Y. Wang, P. Liu, A.-Y. Fu, Y.-N. Zhang, J.-C. Jin and Q.-Z. Shi, Cryst. Growth Des., 2010, 10, 1443–1450 CrossRef CAS.
- A.-L. Cheng, Y. Ma, J.-Y. Zhang and E.-Q. Gao, Dalton Trans., 2008, 1993–2004 RSC.
-
(a) L. Carlucci, G. Ciani, A. Gramaccioli, D. M. Proserpio and S. Rizzato, CrystEngComm, 2000, 2, 154–163 RSC;
(b) Y.-H. Li, C.-Y. Su, A. M. Goforth, K. D. Shimizu, K. D. Gray, M. D. Smith and H.-C. z. Loye, Chem. Commun., 2003, 1630–1631 RSC;
(c) A.-L. Cheng, N. Liu, Y.-F. Yue, Y.-W. Jiang, E.-Q. Gao, C.-H. Yan and M.-Y. He, Chem. Commun., 2007, 407–409 RSC;
(d) A. M. P. Peedikakkal and J. J. Vittal, Cryst. Growth Des., 2008, 8, 375–377 CrossRef CAS.
- V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2004, 6, 378–395 RSC.
-
(a) Y. Ling, L. Zhang, J. Li and A. X. Hu, Cryst. Growth Des., 2009, 9, 2043–2046 CrossRef CAS;
(b) O. Ermer, J. Am. Chem. Soc., 1988, 110, 3747–3754 CrossRef CAS.
- S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460–1494 CrossRef.
-
(a) Y.-F. Hsu, C.-H. Lin, J.-D. Chen and J.-C. Wang, Cryst. Growth Des., 2008, 8, 1094–1096 CrossRef CAS;
(b) H. Kim and M. P. Suh, Inorg. Chem., 2005, 44, 810–812 CrossRef CAS;
(c) L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, Chem.–Eur. J., 2002, 8, 1519–1526 CrossRef CAS;
(d) H.-X. Zhang, Q.-X. Yao, X.-H. Jin, Z.-F. Ju and J. Zhang, CrystEngComm, 2009, 11, 1807–1810 RSC.
- Y.-B. Men, J. Sun, Z.-T. Huang and Q.-Y. Zheng, CrystEngComm, 2009, 11, 978–979 RSC.
-
G. Sheldrick, Program SADABS: Area-Detector Absorption Correction, University of Göttingen, Germany, 1996 Search PubMed.
-
M. Sheldrick, SHELXS-97, SHELXL-97, Programs for Crystal Structure Analysis, University of Göttingen, Germany, 1997 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.