DOI:
10.1039/C0CE00073F
(Communication)
CrystEngComm, 2011,
13, 47-51
Novel form V of tolbutamide and a high Z′ crystal structure of form III†
Received
9th April 2010
, Accepted 10th October 2010
First published on 28th October 2010
Tolbutamide (TBM) is a sulfonylurea drug to control blood sugar levels in patients with type 2 diabetes.1 Four polymorphs of this drug are reported in the literature.2 Recently, the crystal structures of all the four polymorphs were revisited by determining the single crystal X-ray structure of three forms, and the structure of a fourth polymorph was solved by powder X-ray diffraction.3 There has been a report on the selective crystallization of a metastable form IV of TBM in the presence of 2,6-di-O-methyl-β-cyclodextrin.4 Being aware that the presence of co-crystal formers occasionally gives novel drug polymorphs,5 and the fact that nitro group-containing compounds form co-crystals with the urea functional groupvia the strong urea–nitro N–H⋯O synthon,6 we set up the co-crystallization of TBM with p-nitrophenol, p-nitrobenzoic acid and p-phenylenediboronic acid (see Scheme 1) in an attempt to engineer novel co-crystals and discover novel polymorphs.
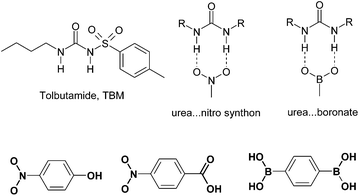 |
| | Scheme 1 The structure of tolbutamide, urea⋯nitro and urea⋯B(OH)2 synthons. Hydrogen bonding donor and acceptor groups in the co-formers are shown in bold. | |
When TBM was co-crystallized with p-nitrophenol and p-phenylenediboronic acid (p-NP and p-PDBA hereafter) in EtOH solvent,‡ we obtained single crystals that solved to give a new crystal structure of TBM with 3 molecules in the asymmetric unit of a P21/n space group (100 K data set, Fig. 1). When reflections were collected on the same crystal at 298 K, the structure solved with Z′ = 1 but now the terminal carbon atoms of the butyl chain were disordered. The room temperature structure (Table 1) matched with that of form III in a recent X-ray diffraction study.3 The unique b-axis was tripled in size the 100 K data set compared to the 298 K measurement (27.189(4), 9.043(4) Å). A close view of the reciprocal lattice by the RLATT program showed reflections for the triple-sized unit cell (Fig. S1†). A similar unit cell compared to the room temperature data can be visualized in the 100 K reflections by removing two rows of reflections, but this small unit cell structure did not refine properly (R1 = 0.1125, GoF = 1.114). On the other hand, the correctly solved and refined crystal structures had a lower R-factor and a better GoF (0.0652, 0.963 at 100 K; 0.0714, 1.099 at 298 K). Both the 100 K and the room temperature structures have essentially the same packing arrangements of molecules, except for a small difference in the orientation of the butyl chain terminal carbon atoms in one of the three symmetry-independent molecules (Fig. 2, molecular ORTEP plots are shown in Fig. S2†). The main synthon in both structures is the urea tape motif, further stabilized by N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S hydrogen bonds (the metrics are listed in Table 2). We believe that p-NP and p-PDBA play a role in producing the high Z′ modulated structure7 of TBM form III because known form I crystallized when the additive was absent. The main lines of the calculated X-ray powder diffraction (XRPD) patterns of the Z′ = 1 and Z′ = 3 structures overlay peak-to-peak and hence they are called modulated structures.7 The similarity of the urea⋯boronate hydrogen bonding8 to that of the urea⋯nitro6 synthon (Scheme 1) is a possible reason for these two molecules giving the same polymorphic result. However, we were unable to detect even a trace of co-crystal formation, as indicated by changes in IR, NIR and Raman frequencies, and XRPD lines.
S hydrogen bonds (the metrics are listed in Table 2). We believe that p-NP and p-PDBA play a role in producing the high Z′ modulated structure7 of TBM form III because known form I crystallized when the additive was absent. The main lines of the calculated X-ray powder diffraction (XRPD) patterns of the Z′ = 1 and Z′ = 3 structures overlay peak-to-peak and hence they are called modulated structures.7 The similarity of the urea⋯boronate hydrogen bonding8 to that of the urea⋯nitro6 synthon (Scheme 1) is a possible reason for these two molecules giving the same polymorphic result. However, we were unable to detect even a trace of co-crystal formation, as indicated by changes in IR, NIR and Raman frequencies, and XRPD lines.
| Polymorph |
Form III |
Form V |
|
Complete crystal data for all the polymorphs is given in the ESI.†
|
| Chemical formula |
C12 H18 N2 O3 S |
C12 H18 N2 O3 S |
C12 H18 N2 O3 S |
| Formula weight |
270.34 |
270.34 |
270.34 |
| Space group |
P21/n |
P21/n |
Pbcn
|
|
T/K |
100 |
298 |
298 |
|
a/Å |
11.5613(17) |
11.787(5) |
15.851(6) |
|
b/Å |
27.189(4) |
9.043(4) |
9.288(4) |
|
c/Å |
13.556(2) |
13.955(6) |
19.691(8) |
|
α (°) |
90 |
90 |
90 |
|
β (°) |
102.803(2) |
104.644(7) |
90 |
|
γ (°) |
90 |
90 |
90 |
|
Z/Z′ |
12/3 |
4/1 |
8/1 |
|
V/Å3 |
4155.2(11) |
1439.2(10) |
2899.2(19) |
|
D
c/g cm−3 |
1.296 |
1.248 |
1.239 |
|
R
1[I > 2σ(I)] |
0.0652 |
0.0714 |
0.0784 |
| Gof |
0.963 |
1.099 |
1.062 |
Table 2
Hydrogen bonds in polymorphs III and V.a
| D–H⋯A |
H⋯A/Å |
D⋯A/Å |
< D–H⋯A (°) |
Symmetry code |
|
N–H distance is neutron normalized to 1.009 Å.
|
| Form III (100 K data) |
| N1–H1⋯O9 |
1.80 |
2.773(3) |
161.6 |
1 + x, y, z |
| N2–H2⋯O8 |
2.13 |
2.907(3) |
132.4 |
1 + x, y, z |
| N2–H2⋯O9 |
2.19 |
3.075(3) |
145.7 |
1 + x, y, z |
| N3–H3A⋯O3 |
1.77 |
2.764(4) |
169.7 |
½ + x, ½ − y, −½ + z |
| N4–H4A⋯O2 |
2.06 |
2.893(4) |
138.6 |
½ + x, ½ − y, −½ + z |
| N4–H4A⋯O3 |
2.41 |
3.263(4) |
141.9 |
½ + x, ½ − y, −½ + z |
| N5–H5A⋯O6 |
1.78 |
2.781(4) |
168.9 |
1 − x, −y, 1 − z |
| N6–H6A⋯O5 |
2.05 |
2.871(3) |
137.5 |
1 − x, −y, 1 − z |
| N6–H6A⋯O6 |
2.35 |
3.209(4) |
141.9 |
1 − x, −y, 1 − z |
| Form III (298 K data) |
| N1–H1⋯O3 |
1.79 |
2.770(4) |
163.8 |
![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) − x, −½ + y, ½ − z − x, −½ + y, ½ − z |
| N2–H2⋯O1 |
2.14 |
2.946(4) |
135.3 |
− x, −½ + y, ½ − z |
| N2–H2⋯O3 |
2.22 |
3.094(4) |
143.4 |
− x, −½ + y, ½ − z |
| Form V (298 K data) |
| N2–H1⋯O1 |
2.22 |
3.047(6) |
138.7 |
½ − x, ½ + y, z |
| N2–H1⋯O3 |
2.29 |
3.145(6) |
141.2 |
½ − x, ½ + y, z |
| N1–H2⋯O3 |
1.84 |
2.797(6) |
157.7 |
½ − x, ½ + y, z |
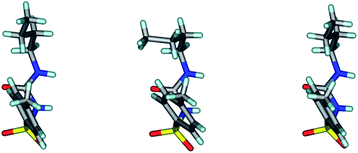 |
| | Fig. 1 The three crystallographic molecules of TBM differ in butyl chain conformation but the rest of the molecule overlays nicely. The middle molecule has a gauche conformation, whereas the two side molecules have an anti conformation. | |
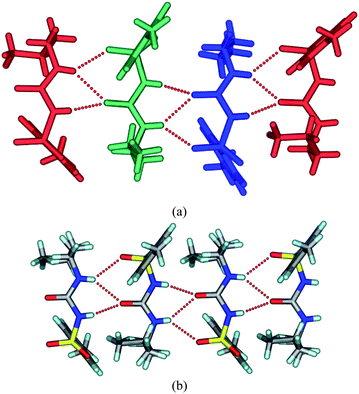 |
| | Fig. 2 (a) The crystal structure of TBM at 100 K. The three symmetry-independent molecules form a urea tape-like 1D chain structure along the b-axis. The terminal atoms of the butyl chain in two molecules are in an anti conformation (green, blue) and one is in a gauche conformation (red). (b) The urea tape motif in the room temperature structure. The butyl chain terminal atoms are in a gauche conformation. | |
Further efforts in this direction focused on the co-crystallization of TBM with p-nitrobenzoic acid because the latter molecule has a stronger hydrogen bonding COOH group compared to p-NP or p-PDBA, and hence may result in a co-crystal. Disappointingly, the result was known TBM form IV.§ Co-crystal formers that can potentially hydrogen bond with the drug5 are believed to play a role in producing a new crystal form9 or a high Z′ structure.10 However, the exact mechanism in such additive- or coformer-induced crystallization of new polymorphs has been delineated in only two systems to our judgement.11 Problems of reproducibility do occur, even after careful experimentation.12
There have been reports of sulfonylurea interacting with nitrate ions.13 The crystallization of TBM in methanol by adding a trace of conc. HNO3 and allowing slow evaporation gave single crystals after 2 d that solved and refined in a Pbcn space group. As such, this is a new space group compared to the reported crystal structures of TBM3 (form I Pna21, form II Pc, form III P21/n, form IV P21/c), an indication that we have crystallized a new polymorph, hereafter referred to as form V. Crystals of form V were very unstable and converted to form I within a few hours. X-Ray reflections were collected at room temperature because the crystal picked up moisture far more rapidly at 100 K and immediately transformed to form I. Attempts at intermediate temperature data collection also gave the same problem. The main synthon in this crystal structure is again the urea tape synthon (N–H⋯OC: 1.84 Å, 157.7°; 2.29 Å, 141.2°) along the b-axis. The SO group also engages in an N–H⋯O hydrogen bond (2.22 Å, 138.7°) to stabilize the linear tape. A C–H⋯OS hydrogen bond (2.76 Å, 132.0°) connects two such tapes, and the overall structure is stabilized by close packing (Fig. 3). Unlike the other three polymorphs (I, II and III), the tolyl group and the butyl chain are on the same side of the urea tape in this structure, similar to that in form IV.
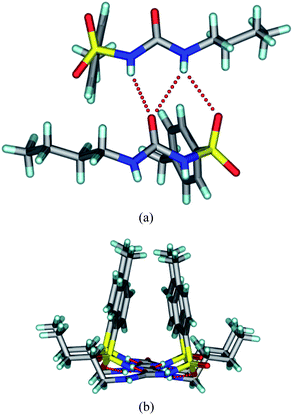 |
| | Fig. 3 (a) The urea tape structure sustained by N–H⋯O hydrogen bonds in the new form V of tolbutamide. (b) The tolyl groups and butyl chains reside on the same side of the urea tape motif. | |
The XRPD patterns of form IV and V calculated from the crystal structures were compared (Fig. 4). The similarity of the 1D urea tape motif and the closeness of the diffraction lines suggests a degree of similarity between these two structures. The molecular overlay in Mercury14 showed a good superposition, but only for 6 out of 15 molecules at the default settings of angle and distance tolerance of 20% between the structures being compared. However, when these tolerances were relaxed to 40% (to include more molecules in the overlay), the number of molecules being superposed increased to 10 out of 15. The differences between the form IV and V structures now showed up clearly (Fig. 5). A good overlay of 15 nearest neighbour molecules (based on the close packing principle that any molecule is surrounded by 12 near neighbour molecules) is generally taken as an indicator of similarity between the crystal structures. The main structural difference between form IV and V is in the direction of the two consecutive urea tapes. The two structures are 1D isostructural (the same urea tape of molecules in similar conformations) but the difference lies in the direction of the urea tape in the two polymorphs (as detailed in the caption of Fig. 6). Finally, the Hirshfeld plots15 of the two polymorphs are also quite different (Fig. S3†).
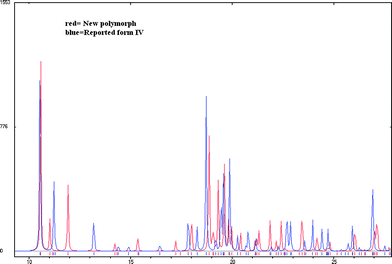 |
| | Fig. 4 There are visible differences in the XRPDs of the recently reported form IV (structure determined by powder data) and the new form V (in this paper). | |
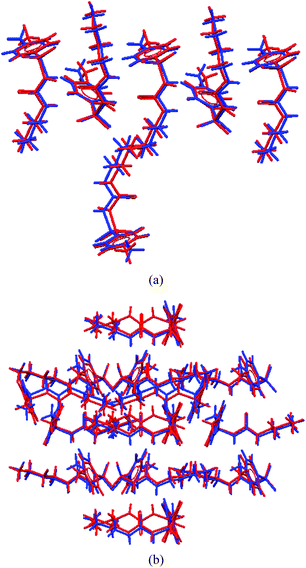 |
| | Fig. 5 (a) An overlay of 6 out of 15 near neighbour molecules at the default setting of 20% variance in bond angles and distances. The 6 molecule superposition is identical. (b) However, when the threshold is increased to 40%, the number of molecules in the overlay increases to 10 out of 15 but the differences in molecular superposition show up clearly. Form V = blue; form IV = red. | |
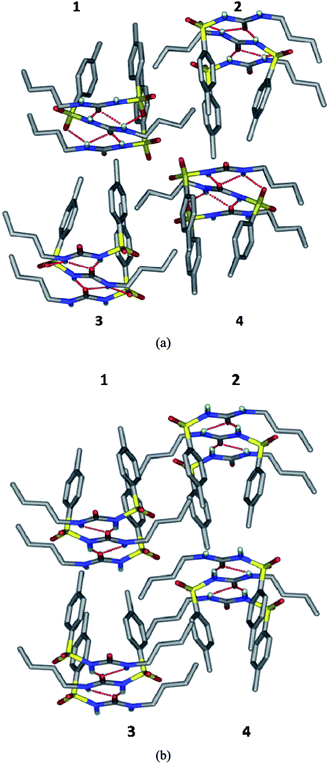 |
| | Fig. 6 The difference in the direction of the urea tape in form V compared to form IV of tolbutamide. A molecular cluster of four adjacent urea tapes is shown in both cases (1, 2, 3 and 4). (a) In polymorph V, the direction of urea tapes 1 and 2 are the same (carbonyl pointing away from the reader) but opposite for 3 and 4 (carbonyl group pointing towards the reader). (b) In form IV, urea tapes 1 and 3 are in the forward direction but 2 and 4 point away. | |
The new form V of tolbutamide was characterized by FT-IR. The N–H stretching (3346 cm−1) of form V is significantly different, and the SO symmetric and asymmetric stretching frequencies (1345 and 1166 cm−1) are slightly different compared to the values reported for polymorphs I–IV (Fig. S4†). The phase change from metastable form V to stable form I at room temperature was monitored by IR spectroscopy (Fig. 7). The microscopy images of form V crystals showed their transformation to form I at room temperature within 2 h (Fig. 8). The transformation of form V to form I was monitored by diagnostic IR peaks.
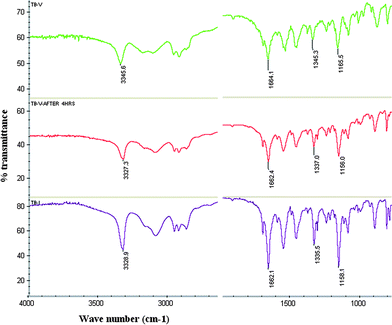 |
| | Fig. 7
FT-IR spectra of TBM form V (green), form V after 4 h at room temperature (red) and form I (magenta) confirm the metastable to stable polymorph phase transformation. | |
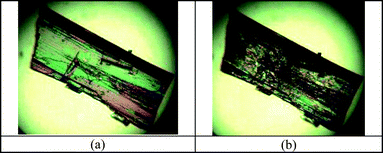 |
| | Fig. 8 (a) TBM form V crystals at room temperature. (b) The transformation of the same crystals to form I after 24 h. The transformation was monitored by FT-IR. | |
In conclusion, serendipity led to the discovery of a novel polymorph of tolbutamide during co-crystallization. Whether the co-former molecule acts as a template in the crystallization of the high Z′ structure of form III is a hypothesis that needs to be verified (a putative scheme is given in Fig. S5†). Known form I was obtained in the absence of an additive. The role of tailored additives and structural mimics in the selective growth of metastable polymorphs is well known.16 This subject has now undergone a renaissance, thanks to the rapid growth of co-crystals,17 leading to the accidental discovery of new polymorphs. The search for novel drug polymorphs in the metastable zone is important because metastable forms generally have an enhanced solubility and bioavailability compared to their stable modifications.18 Hence, there is a need for methods to crystallize metastable polymorphs of drugs and also to stabilize them in the crystal lattice.
N. K. N. thanks the UGC for a fellowship. We thank the DST for research funding (SR/S1/RFOC-01/2007 and SR/S1/OC-67/2006), and the DST (IRPHA) and UGC (PURSE grant) for providing instrumentation and infrastructure facilities.
Notes and references
-
M. J. Thomas and J. A. Thomas in Modern Pharmacology with Clinical Applications, ed. C. R. Craig and R. E. Stitzel, Little Brown & Company, Boston, 5th edn, 1997, pp. 763–776; http://www.drugbank.ca/drugs/DB01124 Search PubMed.
- K. Kimura, F. Hiryama and K. Uekama, J. Pharm. Sci., 1999, 88, 385 CrossRef CAS; E. L. Rowe and B. D. Anderson, J. Pharm. Sci., 1984, 73, 1673 CrossRef CAS; K. A. Nirmala and D. S. S. Gowda, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 1597 CrossRef; J. D. Donaldson, J. R. Leary, S. D. Ross, M. J. K. Thomas and C. H. Smith, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 2245 CrossRef; G. Hasegawa, T. Komasaka, R. Bando, Y. Yoshihashi, E. Yonemochi, K. Fujii, H. Uekusa and K. Terada, Int. J. Pharm., 2009, 369, 12 CrossRef CAS.
- S. Thirunahari, S. Aitipamula, P. S. Chow and R. B. H. Tan, J. Pharm. Sci., 2010, 99, 2975 CAS.
- Y. Sonoda, F. Hirayama, H. Arima, Y. Yamaguchi, W. Saenger and K. Uekama, Cryst. Growth Des., 2006, 6, 1181 CrossRef CAS.
- P. Vishweshwar, J. A. McMahon, M. Oliveira, M. L. Peterson and M. J. Zaworotko, J. Am. Chem. Soc., 2005, 127, 16802 CrossRef CAS; B. Lou, D. Boström and S. P. Velaga, Cryst. Growth Des., 2009, 9, 1254 CrossRef CAS; N. J. Babu, L. S. Reddy, S. Aitipamula and A. Nangia, Chem.–Asian J., 2008, 3, 1122 CrossRef CAS.
- L. S. Reddy, S. K. Chandran, S. George, N. J. Babu and A. Nangia, Cryst. Growth Des., 2007, 7, 2675 CrossRef CAS; M. C. Etter, Z. Urbaňczyk-Lipkowska, M. Zia-Ebrahimi and T. W. Panunto, J. Am. Chem. Soc., 1990, 112, 8415 CrossRef CAS.
- X. Hao, M. A. Siegler, S. Parkin and C. P. Brock, Cryst. Growth Des., 2005, 5, 2225 CrossRef CAS; S. K. Chandran and A. Nangia, CrystEngComm, 2006, 8, 581 RSC; J. W. Steed, CrystEngComm, 2003, 5, 169 RSC.
- Q. Li, F. Xue and T. C. W. Mak, Inorg. Chem., 1999, 38, 4142 CrossRef CAS.
- G. M. Day, A. V. Trask, W. D. S. Motherwell and W. Jones, Chem. Commun., 2006, 54 RSC; M. Wenger and J. Bernstein, Mol. Pharmaceutics, 2007, 4, 355 CrossRef CAS; X. Mei and C. Wolf, Cryst. Growth Des., 2004, 4, 1099 CrossRef CAS; S. Ahn, F. Guo, B. M. Kariuki and K. D. M. Harris, J. Am. Chem. Soc., 2006, 128, 8441 CrossRef CAS.
- M. Rafilovich and J. Bernstein, J. Am. Chem. Soc., 2006, 128, 12185 CrossRef CAS.
- P. K. Thallapally, R. K. R. Jetti, A. K. Katz, H. L. Carrell, K. Singh, K. Lahiri, S. Kotha, R. Boese and G. R. Desiraju, Angew. Chem., Int. Ed., 2004, 43, 1149 CrossRef CAS; R. W. Lancaster, P. G. Karamertzanis, A. T. Hulme, D. A. Tocher, T. C. Lewis and S. L. Price, J. Pharm. Sci., 2007, 96, 3419 CrossRef CAS.
- K. Guo, G. Sadiq, C. Seaton, R. Davey and Q. Yin, Cryst. Growth Des., 2010, 10, 268 CrossRef CAS.
- L. Dupont, O. Dideberg and J. Delarge, Cryst. Struct. Commun., 1981, 10, 89 CAS; L. Dupont, K. Lewinski, K. Stadnicka and J. Delarge, Cryst. Struct. Commun., 1981, 10, 925 CAS; L. Dupont, O. Dideberg, J. Delarge, G. Dive and L. Thunus, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1495 CrossRef.
-
Mercury is a crystal structure visualization and display program distributed by the Cambridge Crystallographic Data Center (CCDC) with the Database package (http://www.ccdc.cam.ac.uk) Search PubMed; another CCDC product, the COSET program, may also be used for crystal structure overlay Search PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19 RSC; J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814 RSC.
- I. Weissbuch, L. Addadi, M. Lahav and L. Leiserowitz, Science, 1991, 253, 637 CAS; R. J. Davey, N. Blagden, G. D. Potts and R. Docherty, J. Am. Chem. Soc., 1997, 119, 1767 CrossRef CAS.
- S. L. Childs and M. J. Zaworotko, Cryst. Growth Des., 2009, 9, 4208 CrossRef CAS (virtual special issue on co-crystals).
- M. Pudipeddi and A. T. M. Serajuddin, J. Pharm. Sci., 2005, 94, 929 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and crystallographic details, RLATT, ORTEP and Hirshfeld plots, and the IR spectra of polymorphs. CCDC reference numbers 772453–772455. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0ce00073f |
‡ Crystallization of form III: A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of tolbutamide and the coformer (p-phenylenediboronic acid, p-nitrophenol or p-nitrobenzoic acid) were ground in a mortar-pestle for 30 min and the resulting solid crystallized from absolute EtOH in separate experiments. TBM form III was obtained in the first and second experiments whereas form IV crystallized in the third experiment. Form V: 0.5 ml of conc. HNO3 was added to 10 ml of MeOH and cooled to −20 °C for 30 min. 30 mg of tolbutamide was dissolved in 10 ml of MeOH and cooled to −20 °C in a separate flask. 1 mL of the acidified MeOH solution was added to the cooled tolbutamide solution and the flask was left undisturbed for crystallization at room temperature. Crystals of from V appeared after 2 days. See Supplementary Information for detailed Experimental. :1 molar ratio of tolbutamide and the coformer (p-phenylenediboronic acid, p-nitrophenol or p-nitrobenzoic acid) were ground in a mortar-pestle for 30 min and the resulting solid crystallized from absolute EtOH in separate experiments. TBM form III was obtained in the first and second experiments whereas form IV crystallized in the third experiment. Form V: 0.5 ml of conc. HNO3 was added to 10 ml of MeOH and cooled to −20 °C for 30 min. 30 mg of tolbutamide was dissolved in 10 ml of MeOH and cooled to −20 °C in a separate flask. 1 mL of the acidified MeOH solution was added to the cooled tolbutamide solution and the flask was left undisturbed for crystallization at room temperature. Crystals of from V appeared after 2 days. See Supplementary Information for detailed Experimental. |
| § See ref. 3 for crystallographic data on forms I–IV of tolbutamide. |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.