Crystal growth on self-assembled monolayers
Aniruddh
Singh
,
In Sung
Lee
,
Kitae
Kim
and
Allan S.
Myerson
*
Department of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue., Cambridge, MA-02139, USA. E-mail: myerson@mit.edu
First published on 2nd November 2010
Abstract
This paper illustrates recent work in the field of crystal growth on self-assembled monolayers (SAMs). The ability to functionalize a surface with the desired properties and their highly ordered structure makes self-assembled monolayers attractive templates for nucleation and crystal growth. These properties could be surface chemistry that leads to specific intermolecular interactions with the nucleating plane of crystals thereby controlling their morphology and crystal form, hydrophobicity and hydrophilicity to create patterned bifunctional surfaces or chirality to create chiral surfaces. Our research group has used SAMs to control crystal morphology and for resolution of racemates. We have also used a SAMs based method to study the effect of the supersaturation generation rate on polymorph obtained, the effect of pH on polymorphic outcome, concomitant nucleation and for the creation of nano sized organic crystals.
 Aniruddh Singh | Aniruddh Singh received his BE in Chemical Engineering in 2004 from the University of Pune, India and his PhD, also in Chemical Engineering, in 2010 from the Illinois Institute of Technology, Chicago under the guidance of Dr Allan S. Myerson. He is currently a Post-Doctoral Research Associate with Dr Bernhardt L. Trout at the Massachusetts Institute of Technology. His research interests include concomitant nucleation, polymorphism, continuous crystallization and racemate resolution using chiral self-assembled monolayers. |
 In Sung Lee | In Sung Lee received his BE in Industrial Chemistry in 1998 from the Hanyang University located in Seoul, South Korea and worked for Samsung SDI Co. as a Research Engineer from 1998 to 2000. He then moved to the Illinois Institute of Technology where he obtained his PhD in Chemical Engineering in 2006 under the guidance of Dr Allan S. Myerson. He is currently employed with SK Holdings Life Science, CMS Division, South Korea. His research interests include concomitant nucleation, polymorph screening and nanosized organic crystals using self-assembled monolayers. |
 Kitae Kim | Kitae Kim received his BE in Chemical Engineering in 2004 from the Sogang University in Seoul, South Korea and PhD in Chemical Engineering in 2010 from the Illinois Institute of Technology, Chicago under the guidance of Dr Allan S. Myerson. He is currently a Post-Doctoral Research Associate with Dr Bernhardt L. Trout at the Massachusetts Institute of Technology. His research interests include concomitant nucleation, polymorphism, solubility, and nanosized organic molecular crystals using self-assembled monolayers. |
 Allan S. Myerson | Allan S. Myerson is Professor of the Practice of Chemical Engineering in the Department of Chemical Engineering at the Massachusetts Institute of Technology, Cambridge, MA. Professor Myerson was educated at Columbia University (BS) and the University of Virginia (MS and PhD). Professor Myerson's research focuses on separations processes in the chemical and pharmaceutical industry with an emphasis on crystallization from solution. He has published 5 books, including the “Handbook of Industrial Crystallization”, 150 papers and is the inventor on 32 US patents. Professor Myerson serves as associate editor of “Crystal Growth and Design” a journal published by the ACS. |
Introduction
Self-assembled monolayers (SAMs) are ordered molecular assemblies that are formed spontaneously by the adsorption of a surfactant with a specific affinity of its head group to a substrate.1 A SAM molecule consists of a head group, chain or backbone and an end group. SAMs are highly ordered and oriented and can incorporate a wide range of groups both in the backbone and as the end group. Therefore they can be used to functionalize a substrate and give it specific properties with fine chemical control.2 The field of self-assembly began3 in the year 1946 when Bigelow, Pickett and Zisman reported that certain types of polar organic molecules are adsorbed from solutions in non-polar solvents to form well-oriented monolayers on polished solid surfaces. They also concluded that the mechanism of the formation of these films on platinum was reversible adsorption from solution and that it definitely was not the accumulation of insoluble films floating at the liquid-air interface as in the Langmuir–Blodgett method.4 Langmuir–Blodgett (LB) films are prepared by transferring Langmuir films (which consist of amphiphilic molecules spread on a liquid surface like water) onto a solid substrate while the most common method to prepare SAMs is by deposition from solution. In principle, a properly cleaned substrate simply has to be dipped into the corresponding solution for a certain period of time for the monolayer to assemble.5 Although the field of self-assembly began in the 1940s, the formation of covalently bound silane monolayers on surfaces exposing free OH groups by Sagiv6 and the identification of the thiol/Au route for SAMs formation by Nuzzo and Allara7(both in the 1980s) increased the popularity of SAMs immensely. Many self-assembly systems have since been studied however monolayers of alkanethiolates on gold are probably the most common3 while silane based SAMs are the second most popular system.1 The wide choice of endgroups that can be anchored to the substrate results in great flexibility in the formation of SAMs and has led to a broad range of applications including the use of SAMs as templates for nucleation and crystal growth.Templated nucleation offers the advantages of lower activation energy of nucleation compared to homogeneous methods, control over crystal morphology,8 control over crystal polymorph9 and control over crystal size.10 Self assembled monolayers have been used as templates for controlled nucleation and crystal growth of biominerals and organic and inorganic crystals. Although SAMs are today widely being studied as templates for nucleation, the early pioneering work involving the use of monolayers as templates was based on Langmuir monolayers.11–14 Landau et al. were the first to report the transfer of structural information from Langmuir monolayers to three-dimensional glycine crystals. In their work they used chiral Langmuir monolayers comprising of alpha-amino acids to induce glycine to crystallize with its {010} face attached to an R amino-acid monolayer while the corresponding S amino-acid monolayer induced attachment of the {0 ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0} face.11 In another significant study, Mann et al. used Langmuir monolayers of stearic acid for the controlled crystallization of CaCO3 from supersaturated solutions. Crystallization in the absence of a monolayer resulted in rhombohedral calcite crystals while the presence of an organized monolayer gave rise to oriented vaterite formation.12 Although Langmuir monolayers were able to induce oriented growth of crystals, it was observed that the specificity of face-selective nucleation was generally not high, and the processes could not be easily controlled because the structures of these organic surfaces were neither homogeneous nor well-defined.15 The high order of the monolayers, their ease of preparation and the extensive structural background of SAMs of alkanethiols on metals led to an interest in using them as organic surfaces to control oriented growth of crystals.15Silane SAMs have also been used as nucleation templates. In some of the early work involving the use of SAMs as templates for nucleation, Tarasevich et al. used silane SAMs to promote heterogeneous nucleation and growth of iron hydroxide crystals.16 Kuther et al. studied the templated growth of calcite, vaterite and aragonite crystals on self-assembled monolayers of substituted alkylthiols on gold and found that the thiol chain length, the ω-substituent on the thiol and the temperature at which crystallization was carried out, all influenced the crystallization.17 Aizenberg et al. studied the oriented growth of calcite controlled by self-assembled monolayers of functionalized alkanethiols supported on gold and silver and found that SAMs terminated in CO2−, SO3−, PO32−, and OH groups were more active in inducing nucleation, whereas SAMs terminated in N(CH3)3+ and CH3 inhibited nucleation.15 Crystal growth on self assembled monolayers continues to generate significant interest and in this report we summarize some of the recent work in the field.
0} face.11 In another significant study, Mann et al. used Langmuir monolayers of stearic acid for the controlled crystallization of CaCO3 from supersaturated solutions. Crystallization in the absence of a monolayer resulted in rhombohedral calcite crystals while the presence of an organized monolayer gave rise to oriented vaterite formation.12 Although Langmuir monolayers were able to induce oriented growth of crystals, it was observed that the specificity of face-selective nucleation was generally not high, and the processes could not be easily controlled because the structures of these organic surfaces were neither homogeneous nor well-defined.15 The high order of the monolayers, their ease of preparation and the extensive structural background of SAMs of alkanethiols on metals led to an interest in using them as organic surfaces to control oriented growth of crystals.15Silane SAMs have also been used as nucleation templates. In some of the early work involving the use of SAMs as templates for nucleation, Tarasevich et al. used silane SAMs to promote heterogeneous nucleation and growth of iron hydroxide crystals.16 Kuther et al. studied the templated growth of calcite, vaterite and aragonite crystals on self-assembled monolayers of substituted alkylthiols on gold and found that the thiol chain length, the ω-substituent on the thiol and the temperature at which crystallization was carried out, all influenced the crystallization.17 Aizenberg et al. studied the oriented growth of calcite controlled by self-assembled monolayers of functionalized alkanethiols supported on gold and silver and found that SAMs terminated in CO2−, SO3−, PO32−, and OH groups were more active in inducing nucleation, whereas SAMs terminated in N(CH3)3+ and CH3 inhibited nucleation.15 Crystal growth on self assembled monolayers continues to generate significant interest and in this report we summarize some of the recent work in the field.
Self-assembled monolayers (SAMs)
Thiol SAMs on gold
As mentioned above, thiol monolayers on gold are the most common. As gold is reasonably inert, substrates can be manipulated in air with lesser concern for contamination. The organosulfur species formed from the adsorption of a thiol on gold is a gold thiolate [Au–S(CH2)nX] where 0 ≤ n ≤ 21 and X = CH3, CHCH2, CH2OH, COOH etc. The reaction may be considered as an oxidative addition of the S–H bond to the gold surface, followed by a reductive elimination of the hydrogen. The gold-sulfur bond is quite strong, with a homolytic bond strength of ∼ 44 kcal/mol18 and the resulting monolayers are quite stable. The monolayers are typically prepared by immersing the substrate in a dilute solution of the adsorbate. Alkanethiol adsorption onto Au(111) surfaces takes place in two distinct steps.3 The first is a very fast step which takes a few minutes and by the end of which contact angles are close to their limiting values and the thickness of the monolayer reaches 80–90% of its maximum. The second is a slow step which takes a few hours and by the end of which the contact angles and the thickness of the monolayer reach their final values.3 The rate of the first step is strongly dependent on thiol concentration. In one study the authors found that in a 1 mM solution the first step took just 1 min, while it required over 100 min in a 1μm solution.19Silane based SAMs
Silane based SAMs are the second most popular system after thiol monolayers on gold.1 The relevant mechanism of silane-based SAM formation is different from thiol SAMs on gold. According to a general reaction scenario of the monolayer formation,20 the growth of alkylsiloxane monolayers proceeds via the nucleation of ordered islands with vertically aligned hydrocarbon chains.21 The alkylsilanes first become hydrolyzed in solution and form the corresponding silanols.22 The silanol molecules then polymerize and form preordered silanol species.23,24 The silanol species adsorb on the surface of the substrate, where they laterally diffuse and aggregate on a thin water layer.6,21 The adsorbed species initially interact with each other and with the surface viasilanol hydrogen bonds and van der Waals interactions before vertical grafting and lateral cross-linking via Si–O–Si bonds take place.21,25 The amount of water in solution needs to be carefully controlled during the self-assembly process. Incomplete monolayers are formed in the absence of water while excess water leads to facile polymerization in solution and polysiloxane deposition on the surface.3 Temperature of the reactional medium has been seen to be another important parameter. It has been reported that a variation of a few degrees of the silanation solution temperature can have a dramatic effect on the layer hydrophobicity as well as its quality. In the same study, the authors observed that an octadecyltrichlorosilane (OTS) monolayer on a silica surface achieves its optimal quality in 2 min at 18 °C while even a 24 h reaction time did not lead to a satisfactory result at 30 °C.26Other SAM systems
In addition to thiol and silane based SAMs, monolayers of anthracene,27azobenzene,28selenols,29 long-chain n-alkanoic acids,30 long-chain hydroxamic acids,31diacetylene amphiphiles,32 di-n-alkyl sulfides,33 di-n-alkyl disulfides,7 thiophenols,34 mercaptopyridines,35 mercaptoanilines,36thiophenes,37 thiocarbamates,38phosphorylcholine,39dodecyl phosphates,40 cysteines41 and stearate42 monolayers have been prepared.SAMs as templates for nucleation and growth of crystals
Templated nucleation offers the advantages of lower activation energy of nucleation compared to homogeneous methods, control over crystal morphology,8 control over crystal polymorph9 and control over crystal size.10 SAMs have been used as templates for controlled nucleation and crystal growth of biominerals and inorganic and organic crystals. Han and Aizenberg have recently reported a method where a stabilized amorphous phase acts as a versatile reservoir system that can be converted in a highly controlled fashion to a crystalline form upon contacting a specially designed nucleating template in water. Amorphous calcium carbonate (ACC) spherulitic particles were induced to form on a self-assembled monolayer (SAM) of hydroxyl-terminated alkanethiols on a gold surface. The ACC was then stored in a dry atmosphere as a reservoir for ions and was induced to crystallize on command by the introduction of water and a secondary surface that was functionalized with carboxylic acid-terminated SAM. The secondary surface acted as a template for oriented and patterned nucleation.43 Pokroy and Aizenberg showed that self-assembled monolayers (SAMs) that template calcite nucleation, in addition to inducing highly oriented crystal growth, also induce a clear modification of the calcite shape from its equilibrium rhombohedron. They demonstrated that this change in shape originates from the anisotropy of lattice mismatches that develop between the nucleating crystal face and the organic SAM in different directions.44 Toworfe and co-workers studied the nucleation and growth of calcium phosphate on amine-, carboxyl- and hydroxyl-silane self-assembled monolayers.45 Freeman et al. reported simulations of calcium carbonate ordering in contact with self-assembled monolayers. The effect of monolayer properties: ionization; epitaxial matching; charge density; and headgroup orientation on the crystallization process were examined. Their results demonstrated that highly charged surfaces are vital to stimulate ordering and crystallization and that template directed crystallization requires charge epitaxy between both the crystal surface and the monolayer.46 Dressler and Mastai studied the crystallization of L-glutamic acid on a self-assembled monolayer of a phenyalanine derivative. They showed that crystallization of L-glutamic acid on SAMs resulted in stabilization of the metastable R-form of L-glutamic acid. Additionally, crystallization onto SAMs led to crystal growth with preferential orientation along the <111> crystal direction.47 Changes in the morphology of SAMs during templated crystallization have also been studied. Lee et al. recently reported that formation of amorphous calcium carbonate (ACC) on mercaptophenol SAMs results in substantial structural disorder within the monolayers. Significantly, despite this static disorder a preferential face of nucleation is observed for crystallization of calcite from ACC on the SAM surfaces.48 Quist and Kakkar demonstrated that a SAM containing a suitably placed backbone structure complementary to the crystallizing molecules can influence the way 4-hydroxybiphenyl molecules pack in the solid state, leading to its crystallization in a new monoclinic polymorph at the interface.49 Mori et al. observed the polycrystallization process of a naphthyl-substituted diamine derivative (NPD) thin film on a self-assembled monolayer (SAM)-modified indium–tin–oxide (ITO) substrate and found that a fluorine-substituted SAM suppressed the growth of the polycrystalline region.50 OTS (Octadecyltrichlorosilane) self-assembled monolayers (SAMs) on glass wafers were used as substrates for the deposition of titanium dioxide thin films. The functionalized organic surface was shown to be effective for promoting the growth of films from titanic aqueous solutions by the liquid phase deposition method.51 Kuther and Tremel studied the templated crystallization of the three polymorphs of calcium carbonate on self-assembled monolayers of ω-substituted alkylthiols on gold surfaces.52Protein crystallization trials using droplets deposited on the surfaces of SAMs were compared to those for which the droplets were deposited on the surfaces of conventional silanized glass microscope slides. For five distinct proteins examined in the crystallization trials a faster rate of crystallization, a larger crystal size and a broader range of crystallization conditions were observed on the SAMs compared to silanized glass.53 Self-assembled monolayers of (+)-L and (−)-D cysteine on gold substrates were used as resolving auxiliaries in the crystallization of rac-glutamic acid and rac-histidine.54,55SAMs on gold as nucleation planes to modify the morphology of crystals
Our research group has used SAMs as nucleation planes for control of crystal morphology. In our initial work in this area it was demonstrated that SAMs and mixed SAMs of rigid thiols on gold can serve as nucleation planes and modify the morphology of glycine crystals. (Fig. 1)56 Self-assembled monolayers (SAMs) and mixed SAMs of 4′-hydroxy-4-mercaptobiphenyl, 4-(4-mercaptophenyl)pyridine, and their mixed SAMs with 4′-methyl-4-mercaptobiphenyl were prepared on gold (111) surfaces and used as templates for the nucleation and growth of glycine crystals. Glycine nucleated in the α-glycine structure independent of hydroxy or pyridine surface concentration. The crystallographic planes corresponding to the nucleation surfaces, for the different SAM surfaces under study, were determined by interfacial angle measurements. We observed that for nucleation on 100% OH surfaces, the glycine crystallographic plane corresponding to the nucleation is {011}, whereas for the 0 and 50% OH surfaces, the crystallographic plane corresponding to the nucleation surface was a {h0l} face. For 25%, 75%, and 100% surface pyridine concentrations, the crystallographic planes corresponding to the nucleation were {010}, {121}, and {1105}, respectively. These differences are attributed to differences in H-bonding between glycine molecules in the nucleating layer and the SAM surface. As interfacial H-bonding increases, the dipoles of glycine molecules within the crystal become more perpendicular to the SAM surface. The direction of dipoles of glycine molecules that nucleated on a pyridine surface are not as close to the surface normal as those of molecules that nucleated on a hydroxyl surface. This implies that the overall H-bonding interactions between the CO2− and NH3+ groups of the glycine and the hydroxyl groups of the SAMs surface are stronger than those between the NH3+ and the pyridine group.56 We also observed57 that 4-mercaptobenzoic acid SAMs on gold induce glycine to nucleate and grow as rod-shaped particles (Fig. 3). This crystal habit is considerably different than glycine crystals grown from aqueous solution where the typical morphology is bipyramidal with the {011}, {110}, {010} and {120} being the dominant growth faces. The crystal plane corresponding to nucleation for the carboxyl terminated 4-MBA SAM is the {12} reflection. The preferred orientation and crystal habit modification is a result of molecular recognition events at the interface involving the chemical functionalities and the structure of the monolayer surface, and the crystal face. Inspection of the {12 } crystallographic plane shows the surface is rough with amine and carboxyl groups sticking out allowing for hydrogen bonds to be formed between the monolayer surface and crystal face.57 Similarly we found that the L-alanine crystallographic planes corresponding to nucleation were {200}, {020}, and {011} on SAMs of 4′-hydroxy-(4-mercaptobiphenyl), 4′-methyl-(4-mercaptobiphenyl), and 4-(4-mercaptophenyl)pyridine on gold (111) surfaces, respectively.8 The appearance of an unobserved {200} face of L-alanine grown in aqueous solution on SAMs of 4′-hydroxy-(4-mercaptobiphenyl) can be attributed to hydrogen bonds forming between the two surfaces. The {200} surface contains alternating methyl (CH3) and carboxylic groups (COO–) that form N–H⋯O and O⋯H–O hydrogen bonds with the hydroxide group of the monolayer film, ideal for binding with surfaces that can serve as both hydrogen bond donors and acceptors. As a result, the preferential interaction leads to the stabilization and appearance of the {200} face. Crystallization of L-alanine on 4-(4-mercaptophenyl)pyridine surfaces resulted in the {011} face as the plane corresponding to nucleation. The preferential interaction of the monolayer with the {011} face can be attributed to hydrogen bonding at the crystal-monolayer interface. Unlike the other two surfaces where they can serve as both hydrogen bond donors and acceptors (4′-hydroxy-4-mercaptobiphenyl) or solely as H-bond donors (4′-methyl-4-mercaptobiphenyl), the pyridine electron pair at the surface only serve as hydrogen bond acceptors. The binding of the pyridine surface and the {011} plane can be explained by the amino and methyl groups protruding out perpendicular to the plane and forming N– H⋯N and C–H⋯N hydrogen bonds with the SAM surface, respectively. In contrast, the 100% CH3 and 100% OH SAM surfaces do not interact as strongly with the hydrogen bond donating plane.8 In the case of DL-valine (Fig. 2), monolayer surfaces that act as hydrogen bond acceptors (e.g., 4′-hydroxy-(4-mercaptobiphenyl) and 4-(4-mercaptophenyl)-pyridine) induced the racemic crystal to nucleate from the {020} plane whereas the nucleating plane for the 4′-methyl-(4-mercaptobiphenyl) surface was the fast-growing {100} face and the hexagonal platelet crystal is almost oriented perpendicular to the SAM surface, tilting uniformly in one direction, suggesting that in addition to the functional group of the SAM the geometry of the SAM also affects the orientation of the crystal. The specific interactions between the {020} plane and the 4′-hydroxy-(4-mercaptobiphenyl) and 4-(4-mercaptophenyl)-pyridine) surfaces occur because of the strong interfacial hydrogen bonding interactions. On the molecular level, the {020} plane has a very rough surface topography with methyl groups protruding diagonally out of the plane, allowing possible hydrogen bonding opportunities. In the hydroxy-terminated SAMs, C–H⋯O hydrogen bonds are formed, whereas on the pyridine surface, the slow-growing crystal face is linked by C–H⋯N hydrogen bonds.8

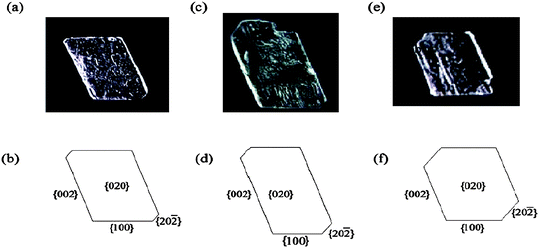 | ||
| Fig. 2 Crystallographic image and morphology of DL-valine crystals grown on (a,b) SAMs of 4-(4-mercaptophenyl)pyridine, (c,d) SAMs of 4′-hydroxy-4-mercaptobiphenyl, and (e,f) SAMs of 4′-methyl-4-mercaptobiphenyl.8 | ||
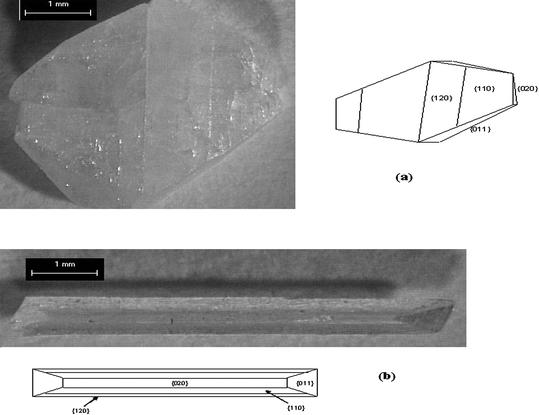 | ||
| Fig. 3 Habit Modification of α-Glycine (a) α-glycine crystal grown in aqueous solution has bipyramidal habit (b) α-glycine crystal on COOH terminated surfaces at moderate supersaturation shows needle-like morphology. | ||
 | ||
| Fig. 4 (a) A patterned substrate having more than 2000 square gold islands with a dimension of 500 μm and (b) crystals of glycine produced on 500 μm islands.79 | ||
 | ||
| Fig. 5 (a) A microscope image showing an array of gold islands with a dimension of 725 μm with ROY crystals on them and (b) different forms of ROY crystallized on 725 μm islands.64 | ||
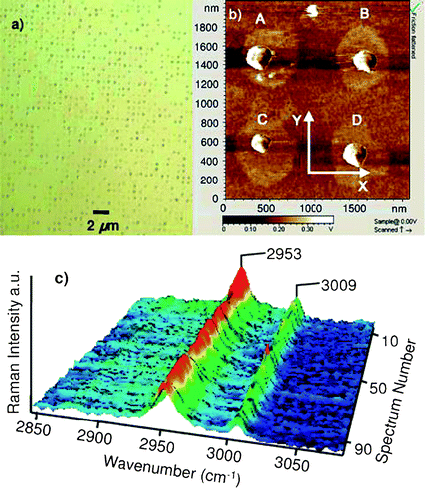 | ||
| Fig. 6 (a) Optical microscope image (×150) of 1600 μm2 and (b) AFM image of 4 μm2 of the 500 nm patterned SAMs on the silicon substrate. The crystals dimensions are: (A) 250 × 220 × 74 nm3, (B) 230 × 205 × 70 nm3, (C) 180 × 160 × 57 nm3, and (D) 220 × 190 × 65 nm3. (c) Raman spectra of 100 individual glycine crystals found on the patterned SAMs.58 | ||
 | ||
| Fig. 7 L-enantiomer crystals obtained on a (a) L-glutathione chiral monolayer and (b) N-acetyl-L-cysteine chiral monolayer. | ||
The observation of crystal nucleation and orientation can be attributed to the strong interfacial interactions, in particular, hydrogen bonding, between the surface functionalities of the monolayer film and the individual molecules of the crystallizing phase. Molecular modeling studies were also undertaken to examine the molecular recognition process across the interface between the surfactant monolayer and the crystallographic planes. Binding energies between SAMs and particular amino acid crystal faces were calculated and the results were in good agreement with the observed nucleation planes of the amino acids. These results suggest that binding energy calculations can be a valid method to screen self-assembled monolayers as potential templates for nucleation and growth of organic and inorganic crystals.
Crystallization on patterned surfaces of SAMs
Another application of SAMs developed in our laboratory is the use of patterned substrates of self-assembled monolayers (SAMs), which consist of lyophilic islands surrounded by a lyophobic domain10,58 which can be used as a probe for studying nucleation. Metallic gold islands are formed by evaporation of titanium through a mesh onto glass slides, followed by a subsequent evaporation of gold through the stencil. The dimensions and patterns of the islands depend on the size and shape of the hole. Typically, the holes are shaped as squares. A hydrophilic monolayer is selected as the thiol monolayer that is self-assembled onto the gold surface, while a hydrophobic monolayer is used to backfill the glass-exposed substrate. The patterned bifunctional surface is immersed and slowly withdrawn from undersaturated solutions. The solution preferentially wets the metallic islands and arrays of small solution droplets are generated. Solvent evaporation from the droplets or vapor diffusion leads to crystallization (Fig. 4). This method allows us to carry out a large number of independent crystallization trials with a minimal amount of material. It is also possible to achieve statistical accuracy of the effect of different crystallization variables on polymorphic outcome by analysis of a large number of islands. Crystallization is confined to the islands and the size and area of the particle can be controlled by varying the feature size of the islands or the concentration of the solution. This technique can be used to generate nanometer size crystals. Additionally, fast solvent evaporation in the drops can lead to high supersaturation, where high energy metastable polymorphs are kinetically favored and can nucleate and grow. Also, given that different polymorphic forms are obtained on the identical islands under the same crystallization conditions, crystallization in constrained environments can aid in understanding the concomitant nucleation of polymorphs. Patterned self-assembled monolayers on metal substrates have also been prepared by microcontact printing with an elastomeric stamp.59,60 Patterned SAMs prepared by this method were used for controlling crystal nucleation and achieving ordered crystallization of calcite from calcium chloride solutions.61To study effect of rate of supersaturation generation on polymorphic outcome
We have studied the effect of the rate of generating supersaturation on polymorphic outcome for glycine,62sulfathiazole and mefenamic acid63 using the patterned substrates of SAMs described above. For glycine, moderately slow evaporation favored the nucleation of α-form crystals while high supersaturation was created with fast evaporation and led to the formation of the less stable β-form. Also, the small volumes produced by the patterned metallic islands minimize the prospect that the metastable form might transform to the more stable form viasolvent-mediated transformation. Similar behavior was observed for sulfathiazole and mefenamic acid with an increase in island size leading to a decrease in the rate of supersaturation generation and an increase in the percentage of islands yielding the stable forms.To study concomitant nucleation
Concomitant nucleation is an interesting phenomenon where different polymorphic forms are obtained under seemingly identical conditions. An interplay between thermodynamic and kinetic factors is believed to result in more than one polymorph being obtained. Patterned substrates of SAMs have been used to study concomitant nucleation.63,64 The large sample sizes in this method allow a higher probability for detection of a polymorphic form having a small Pi, where Pi is the probability of yielding an i-form crystal in a crystallization trial. 5-methyl-2-[(2-nitrophenyl)amino]-3-thiophenecarbonitrile (known as ROY for its red, orange and yellow crystals) is a good model compound to study concomitant nucleation due to a high number of well characterized forms. Crystallization experiments were carried out using patterned substrates of SAMs and solutions of ROY in dimethylsulfoxide (DMSO). We were able to show that increasing the number of crystallization trials could lead to nucleation of polymorphs that have escaped solution crystallization experiments using conventional methods. We were able to crystallize six of the seven stable polymorphs of ROY concomitantly, including form YT04, which to the best of our knowledge, has never been obtained from solution crystallization (Fig. 5). We have also demonstrated how increasing the number of crystallization trials can help crystallize polymorphs which may not be obtained in a fewer number of trials due to statistical reasons.64 Also, both known forms of mefenamic acid and four out of the five known forms of sulfathiazole have been obtained concomitantly using the patterned SAMs method.63To study effect of pH on polymorphic outcome
The simplest amino acid glycine exists in three distinct polymorphic: α,65 β-,66 and γ-forms67 at ambient environment. The α-form is built from centrosymmetric zwitterionic dimers,65 while the β- and γ-forms in non-centrosymmetric structure.68 At room temperature, γ-glycine is the thermodynamically most stable form.69 However, in neutral aqueous solution of pH ca. 6.20, the kinetic α-form is normally obtained.65 Typically, the γ-form is produced from acidic and basic solutions.67,69β-Glycine is the least stable form compared to α- and γ-forms. The metastable α-form of glycine is usually produced from aqueous solution in the pH range between 3.8 and 8.9, and the thermodynamically most stable γ-form is generated from acidic and basic solutions.70 It had been suggested70 that charged glycine species, protonated (+H3NCH2CO2H) at low pH (less than 3.8) and deprotonated (H2NCH2CO2−) at high pH (greater than 8.9), inhibit the growth of α-glycine, enabling the crystallization of the γ-form. Consequently, controlling the precise pH domain (less than 3.8 and greater than 8.9) favored the formation of γ-glycine. However, it had also been demonstrated that γ-glycine can be crystallized in neutral aqueous solutions with additives71,72 or by irradiation of intense polarized laser light,73,74 DC electric field,75 slow evaporation of solvent using an evaporation-based crystallization platform,76 in a microporous membrane,77 and by thin film evaporation of solution on the walls of a glass container.78 We conducted crystallization experiments using patterned substrates of SAMs with aqueous glycine solutions at three different pHs and concentrations. More than 2000 islands per substrate were analyzed to achieve statistical accuracy of the polymorph distribution of crystals formed on the substrate due to the stochastic nature of polymorphism from solution crystallization. The role of pH on the probability of forming each polymorphic modification was investigated and it was found that at solution pHs less than 3.40 and greater than 10.10, the charged glycine species does not appear to entirely inhibit the growth of kinetically favored α-glycine, but reduces the possibility of producing the α-form, thereby increasing the chance of forming γ-glycine in the solution.79To generate nanosized organic molecular crystals
The formation of organic molecular crystals with sizes below 500 nm is of great interest to the pharmaceutical industry since an enhanced solubility and dissolution rate can potentially increase drug bioavailability. Recently, we fabricated bifunctional (hydrophilic and hydrophobic) patterned surfaces with circular shaped islands with diameters of 500 nm using photolithography and SAMs.58 The circular islands were covered with a hydrophilic SAM (3-aminopropyl-triethoxysilane (ATPES)), while the rest of the surface was functionalized with a hydrophobic SAM (octadecyltrichlorosilane (OTS)). The diameter of the solution droplets generated on each hydrophilic island was below 500 nm, and consequently, the crystal size formed on each island could be controlled to be smaller than 500 nm. These patterned engineered surfaces were used for crystallizing glycine with a lateral dimension below 200 nm in a confined volume while controlling supersaturation. Individual crystals were characterized with AFM and Raman spectroscopy (Fig. 6) and determined to be the metastable β-form. The solubility of the crystals in ethanol was measured as a function of crystal size and employed along with the Ostwald-Freundlich equation to predict solubility enhancement vs. crystal size.Chiral SAMs as resolving auxiliaries in the crystallization of racemates
Chiral drugs are a subgroup of drug substances that contain one or more chiral centers. More than one-half of marketed drugs are chiral.80 In some cases, chiral drugs are marketed as racemates as it is frequently costly to resolve a racemic mixture into pure enantiomers. Opposite enantiomers often differ significantly in their pharmacological,81 toxicological,82 pharmacodynamic and pharmacokinetic83,84 properties. From the points of view of safety and efficacy, the pure enantiomer is preferred over the racemate in many marketed dosage forms.85 Thus, resolution of enantiomer mixtures is an active area of research. Chiral surfaces can be created by anchoring or adsorption of a chiral molecule on a nonchiral surface.86–88 The ability to differentiate between enantiomers of a chiral molecule is one of the most interesting properties of a chiral surface. Recently, we used self assembled monolayers of L-cysteine, D-cysteine, N-acetyl-L-cysteine, L-glutathione and D-penicillamine on gold as resolving auxiliaries in the crystallization of the amino acid valine.89 We were able to obtain one enantiomer in excess on the crystals obtained on the chiral SAMs when starting with racemic solutions. The enantiomer obtained in excess had the opposite chirality of the monolayer used. On decreasing the supersaturation the relative intensity of the characteristic XRD peak of the pure enantiomer increased, indicating that chiral resolution was enhanced. However, further experiments need to be carried out to obtain a statistically significant number of samples to confirm these trends. In addition crystals of the pure enantiomer were obtained when starting with a solution having an EE value of 50% (Fig. 7). Control experiments carried out without chiral SAMs resulted in mixtures of the pure enantiomer and racemic compound. The enantiomer obtained on chiral SAMs at conditions of large enantiomeric excess was the one initially present in excess regardless of the chirality of the monolayer being used.Concluding remarks
The stability of self-assembled monolayers and their ability to be formed on a wide variety of surfaces and the wide choice of endgroups that can be anchored to the substrate results in great flexibility in the formation of SAMs and has led to a broad range of applications. The ability to functionalize a surface with the desired properties and their highly ordered structure makes self-assembled monolayers attractive templates for nucleation and crystal growth. These properties could be surface chemistry that leads to specific intermolecular interactions with the nucleating plane of crystals thereby controlling their morphology and crystal form, hydrophobicity and hydrophilicity to create patterned bifunctional surfaces or chirality to create chiral surfaces. Our recent work in the field ranges from the use of SAMs to control crystal morphology and for resolution of racemates to the use of a SAMs based method to study the effect of the supersaturation generation rate on polymorph obtained, the effect of pH on polymorphic outcome, concomitant nucleation and creation of nano sized organic crystals using a SAMs based method. Further work is needed to increase the applications of SAMs as templates for crystallization and to better understand the mechanism behind the effect of SAMs on crystal morphology, polymorphic form and in chiral resolution.References
- F. Schreiber, J. Phys.: Condens. Matter, 2004, 16, R881 CrossRef CAS.
- A. Ulman, S. D. Evans, Y. Shnidman, R. Sharma, J. E. Eilers and J. C. Chang, J. Am. Chem. Soc., 1991, 113, 1499 CrossRef CAS.
- A. Ulman, Chem. Rev., 1996, 96, 1533 CrossRef CAS.
- W. C. Bigelow, D. L. Pickett and W. A. Zisman, J. Colloid Sci., 1946, 1, 513 CAS.
- F. Schreiber, Prog. Surf. Sci., 2000, 65, 151 CrossRef CAS.
- J. Sagiv, J. Am. Chem. Soc., 1980, 102, 92 CrossRef CAS.
- R. G. Nuzzo and D. L. Allara, J. Am. Chem. Soc., 1983, 105, 4481 CrossRef CAS.
- A. Y. Lee, A. Ulman and A. S. Myerson, Langmuir, 2002, 18, 5886 CrossRef CAS.
- R. Hiremath, J. A. Basile, S. W. Varney and J. A. Swift, J. Am. Chem. Soc., 2005, 127, 18321 CrossRef CAS.
- A. Y. Lee, I. S. Lee, S. S. Dette, J. Boerner and A. S. Myerson, J. Am. Chem. Soc., 2005, 127, 14982 CrossRef CAS.
- E. M. Landau, M. Levanon, L. Leiserowitz, M. Lahav and J. Sagiv, Nature, 1985, 318, 353 CrossRef CAS.
- S. Mann, B. R. Heywood, S. Rajam and J. D. Birchall, Nature, 1988, 334, 692 CrossRef CAS.
- E. M. Landau, S. G. Wolf, M. Levanon, L. Leiserowitz, M. Lahav and J. Sagiv, J. Am. Chem. Soc., 1989, 111, 1436 CrossRef CAS.
- I. Weissbuch, F. Frolow, L. Addadi, M. Lahav and L. Leiserowitz, J. Am. Chem. Soc., 1990, 112, 7718 CrossRef CAS.
- J. Aizenberg, A. J. Black and G. M. Whitesides, J. Am. Chem. Soc., 1999, 121, 4500 CrossRef CAS.
- B. J. Tarasevich, P. C. Rieke and J. Liu, Chem. Mater., 1996, 8, 292 CrossRef.
- J. Küther, R. Seshadri, W. Knoll and W. Tremel, J. Mater. Chem., 1998, 8, 641 RSC.
- L. H. Dubois and R. G. Nuzzo, Annu. Rev. Phys. Chern., 1992, 43, 437 Search PubMed.
- C. D. Bain, E. B. Troughton, Y. T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321 CrossRef CAS.
- R. Bautista, N. Hartmann and E. Hasselbrink, Langmuir, 2003, 19, 6590 CrossRef CAS.
- A. N. Parikh, D. L. Allara, I. B. Azouz and F. Rondelez, J. Phys. Chem., 1994, 98, 7577 CrossRef CAS.
- M. E. McGovern, K. M. R. Kallury and M. Thompson, Langmuir, 1994, 10, 3607 CrossRef CAS.
- T. Vallant, J. Kattner, H. Brunner, U. Mayer and H. Hoffmann, Langmuir, 1999, 15, 5339 CrossRef CAS.
- T. Vallant, H. Brunner, U. Mayer, H. Hoffmann, T. Leitner, R. Resch and G. Friedbacher, J. Phys. Chem. B, 1998, 102, 7190 CrossRef CAS.
- M. M. Sung, C. Carraro, O. W. Yauw, Y. Kim and R. Maboudian, J. Phys. Chem. B, 2000, 104, 1556 CrossRef CAS.
- P. Silberzan, L. Leger, D. Ausserre and J. J. Benattar, Langmuir, 1991, 7, 1647 CrossRef CAS.
- I. G. Hill, J. Hwang, A. Kahn, C. Huang, J. E. McDermott and J. Schwartz, App. Phys. Lett., 2007, 90, 0121091 Search PubMed.
- G. Pace, V. Ferri, C. Grave, M. Elbing, C. von Hanisch, M. Zharnikov, M. Mayor, M. A. Rampi and P. Samori, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9937 CrossRef CAS.
- M. G. Samant, C. A. Brown and J. G. Gordon, Langmuir, 1992, 8, 1615 CrossRef CAS.
- N. E. Schlotter, M. D. Porter, T. B. Bright and D. L. Allara, Chem. Phys. Lett., 1986, 132, 93 CrossRef.
- J. P. Folkers, C. B. Gorman, P. E. Laibinis, S. Buchholz, G. M. Whitesides and R. G. Nuzzo, Langmuir, 1995, 11, 813 CrossRef CAS.
- D. Y. Huang and Y.-T. Tao, Bull. Inst. Chem., Acad. Sin., 1986, 33, 73 Search PubMed.
- E. B. Troughton, C. D. Bain, G. M. Whitesides, R. G. Nuzzo, D. L. Allara and M. D. Porter, Langmuir, 1988, 4, 365 CrossRef CAS.
- E. Sabatani, J. Cohen-Boulakia, M. Bruening and I. Rubinstein, Langmuir, 1993, 9, 2974 CrossRef CAS.
- M. A. Bryant, S. L. Joa and J. E. Pemberton, Langmuir, 1992, 8, 753 CrossRef CAS.
- W. Hill and B. Wehling, J. Phys. Chem., 1993, 97, 9451 CrossRef CAS.
- S. K. Arya, P. R. Solanki, S. P. Singh, K. Kaneto, M. K. Pandey, M. Datta and B. D. Malhotra, Biosens. Bioelectron., 2007, 22, 2516 CrossRef CAS.
- J. A. Mielczarski and R. H. Yoon, Langmuir, 1991, 7, 101 CrossRef CAS.
- S. Chen, J. Zheng, L. Li and S. Jiang, J. Am. Chem. Soc., 2005, 127, 14473 CrossRef CAS.
- C. S. Tang, M. Antoni, I. Schonbachler, B. Keller, M. Textor and J. Voros, ECS Trans., 2005, 28, 29 Search PubMed.
- J. M. Cooper, K. R. Greenough and C. J. McNeil, J. Electroanal. Chem., 1993, 347, 267 CrossRef CAS.
- P. Fenter and N. C. Sturchio, Geochim. Cosmochim. Acta, 1999, 63, 3145 CrossRef CAS.
- T. Y. Han and J. Aizenberg, Chem. Mater., 2008, 20, 1064 CrossRef CAS.
- B. Pokroy and J. Aizenberg, Crys. Eng. Comm., 2007, 9, 1219 Search PubMed.
- G. K. Toworfe, R. J. Composto, I. M. Shapiro and P. Ducheyne, Biomaterials, 2006, 27, 631 CrossRef CAS.
- C. L. Freeman, J. H. Harding and D. M. Duffy, Langmuir, 2008, 24, 9607 CrossRef CAS.
- D. H. Dressler and Y. Mastai, Cryst. Growth Des., 2007, 7, 847 CrossRef CAS.
- J. R. I. Lee, T. Y. Han, T. M. Willey, D. Wang, R. W. Meulenberg, J. Nilsson, P. M. Dove, L. J. Terminello, T. van Buuren and J. J. De Yoreo, J. Am. Chem. Soc., 2007, 129, 10370 CrossRef CAS.
- F. Quist and A. Kakkar, J. Colloid Interface Sci., 2007, 313, 378 CrossRef CAS.
- T. Mori, S. Nishino, T. Nishikawa and S. Ogawa, Org. Electron., 2008, 9, 63 CAS.
- Z. L. He, Z. Yu, H. Miao, G. Q. Tan and Y. Liu, Sci. China, Ser. E: Technol. Sci., 2009, 52, 137 Search PubMed.
- J. Kuther and W. Tremel, Thin Solid Films, 1998, 327–329, 554 CrossRef CAS.
- T. Pham, D. Lai, D. Ji, W. Tuntiwechapikul, J. M. Friedman and T. R. Lee, Colloids Surf., B, 2004, 34, 191 CrossRef CAS.
- D. H. Dressler and Y. Mastai, Chirality, 2007, 19, 358 CrossRef CAS.
- D. H. Dressler and Y. Mastai, J. Colloid Interface Sci., 2007, 310, 653 CrossRef CAS.
- J. F. Kang, J. Zaccaro, A. Ulman and A. S. Myerson, Langmuir, 2000, 16, 3791 CrossRef CAS.
- A. Y. Lee, Ph.D. Thesis, Illinois Institute of Technology, 2005.
- K. Kim, I. S. Lee, A. Centrone, T. A. Hatton and A. S. Myerson, J. Am. Chem. Soc., 2009, 131, 18212 CrossRef CAS.
- A. Kumar, N. L. Abbott, H. A. Biebuyck, E. Kim and G. M. Whitesides, Acc. Chem. Res., 1995, 28, 219 CrossRef CAS.
- Y. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 550 CrossRef CAS.
- J. Aizenberg, A. J. Black and G. M. Whitesides, Nature, 1999, 398, 495 CrossRef CAS.
- A. Y. Lee, I. S. Lee and A. S. Myerson, Chem. Eng. Technol., 2005, 29, 281.
- I. S. Lee, A. Y. Lee and A. S. Myerson, Pharmacol. Res., 2008, 25, 960 CrossRef CAS.
- A. Singh, I. S. Lee and A. S. Myerson, Cryst. Growth Des., 2009, 9, 1182 CrossRef CAS.
- R. E. Marsh, Acta Crystallogr., 1958, 11, 654 CrossRef CAS.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1905, 38, 2917.
- Y. Iitaka, Proc. Jpn. Acad., 1954, 30, 109 CAS.
- E. S. Ferrari, R. J. Davey, W. I. Cross, A. L. Gillon and C. S. Towler, Cryst. Growth Des., 2003, 3, 53 CrossRef CAS.
- Y. Iitaka, Acta Crystallogr., 1961, 14, 1 CrossRef CAS.
- C. S. Towler, R. J. Davey, R. W. Lancaster and C. J. Price, J. Am. Chem. Soc., 2004, 126, 13347 CrossRef CAS.
- I. Weissbuch, D. Zbaida, L. Addadi, L. Leiserowitz and M. Lahav, J. Am. Chem. Soc., 1987, 109, 1869 CrossRef CAS.
- I. Weissbuch, V. Yu. Torbeev, L. Leiserowitz and M. Lahav, Angew. Chem., Int. Ed., 2005, 44, 3226 CrossRef CAS.
- J. Zaccaro, J. Matic, A. S. Myerson and B. A. Garetz, Cryst. Growth Des., 2001, 1, 5 CrossRef CAS.
- B. A. Garetz, J. Matic and A. S. Myerson, Phys. Rev. Lett., 2002, 89, 175501 CrossRef.
- J. E. Aber, S. Arnold, B. A. Garetz and A. S. Myerson, Phys. Rev. Lett., 2005, 94, 145503 CrossRef.
- G. He, V. Bhamidi, R. B. H. Tan, P. J. A. Kenis and C. F. Zukoski, Cryst. Growth Des., 2006, 6, 1746 CrossRef CAS.
- G. D. Profio, S. Tucci, E. Curcio and E. Drioli, Cryst. Growth Des., 2007, 7, 526 CrossRef CAS.
- M. Xu and K. D. M. Harris, J. Phys. Chem. B, 2007, 111, 8705 CrossRef CAS.
- I. S. Lee, K. Kim, A. Y. Lee and A. S. Myerson, Cryst. Growth Des., 2008, 8, 108 CrossRef CAS.
- J. S. Millership and A. Fitzpatrick, Chirality, 1993, 5, 573 CrossRef CAS.
- M. M. Islamm, J. G. Mahdi and I. D. Bowen, Drug Saf., 1997, 149 CrossRef.
- I. Wainer, 1993. Drug stereochemistry: Analytical methods and pharmacology. New York: Marcel Dekker Search PubMed.
- D. Drayer, Clin. Pharmacol. Ther., 1986, 40, 125 CAS.
- K. M. Midha, G. McKay, M. J. Rawson and J. W. Hubbard, J. Pharm. Sci., 1998, 87, 797 CrossRef CAS.
- I. K. Reddy and R. Mehvar. 2004. Chirality in drug design and development.New York: Marcel Dekker Search PubMed.
- M. O. Lorenzo, C. J. Baddeley, C. Muryn and R. Raval, Nature, 2000, 404, 376 CrossRef CAS.
- M. Schunack, E. Laegsgaard, I. Steengaard, I. Johannsen and F. Besenbacher, Angew. Chem., Int. Ed., 2001, 40, 2623 CrossRef.
- K. H. Ernst, Y. Kuster, R. Fasel, M. Muller and U. Ellerbeck, Chirality, 2001, 13, 675 CrossRef CAS.
- A. Singh and A. S. Myerson, J. Pharm. Sci., 2010, 99(9), 3931–3940 CAS.
| This journal is © The Royal Society of Chemistry 2011 |