Self-assembly and solvent-mediated structural transformation of one-dimensional cluster-based coordination polymer†
Jian-Jun
Zhang
a,
Cynthia S.
Day
b and
Abdessadek
Lachgar
*b
aDepartment of Chemistry, Dalian University of Technology, Dalian, Liaoning 116024, P R China
bDepartment of Chemistry, Wake Forest University, Winston-Salem, North Carolina 27109, USA. E-mail: lachgar@wfu.edu; Fax: (+1) 336-758-4656; Tel: (+1) 336-758-4676
First published on 27th August 2010
Abstract
Reactions between [Nb6Cl12(CN)6]4− and [Mn(salen)]+ in MeOH–H2O solvent mixture led to the formation of a new compound: {[(Mn2(salen)2)][[Mn(salen)(H2O)]0.67[Mn(salen)(MeOH)]1.33(Nb6Cl12(CN)6)]}·1.7H2O·2MeOH (1). Single crystal X-ray analysis has shown that 1 possesses 1D framework built of heterotrimeric supramolecular anions, {[Mn(salen)(s)]2[Nb6Cl12(CN)6]}2− and [Mn2(salen)2]2+ phenoxo bridged dimers linked to each other through two cyanide ligands located in axial positions. When 1 was left in a methanolic solution of (Me4N)+, its 1D framework was transformed into a 2D framework of the previously reported compound [Me4N]2[(Mn(salen))2(Nb6Cl12(CN)6))] (2) which has a 2D anionic 4,4-network built of [Nb6Cl12(CN)6]4− and [Mn(salen)]+ nodes linked by CN ligands (Zhou, Day and Lachgar, Chem. Mater., 2004, 16, 4870).1Magnetic susceptibility measurements indicate that 1 exhibits weak antiferromagnetic interactions due to the phenolate-bridged out-of-plane Mn(III) dimers with J = −1.50(5) cm−1. Thermal stability of 1 was studied.
Introduction
Edge-bridged octahedral metal clusters M6Li12La6 (M = Nb, Ta; L = halide) are characterized by their various electronic and magnetic properties arisen from the presence of metal–metal bond.2 When equipped with suitable terminal ligands, these clusters can self-assemble with different metal ions or metal complexes to form nanomolecular assemblies or hybrid inorganic–organic materials with different topologies and interesting properties.3–6 Compared with mononuclear species, the use of M6clusters as nodes is characterized by their large size (≧1 nm), availability of multiple coordination sites, as well as the potential to create materials with structural features and chemical–physical properties that can be affected by the use of different metals and/or different ligands.3–6Hybrid materials and coordination polymers have received much attention in the past decade due primarily to their potential applications and attractive structures.7 Initial investigations focused on rigid frameworks and recently there has been growing interest in new generation of flexible frameworks bearing pore size adjustment or even giant structural changes in response to external stimuli.8 This interesting dynamic structural transformations can be used to prepare a new class of materials that possess new functions such as switching or sensing.9 Most transformations have been suggested to have a solid state (crystal-to-crystal or amorphous-to-crystal) mechanism, but solvent-mediated mechanism has been proposed for other transformations.10
Previous studies by our group showed that different supramolecular assemblies and multi-dimensional coordination polymers can be prepared through the combination of the niobium cyanochloride clusters [Nb6Cl12(CN)6]4− and Mn(III) complexes of the tetradentate salen-type ligands.1,6Solvent-mediated structural transformation behavior (2D → 2D or 2D → 0D) has been demonstrated for some of these compounds.6 Herein we report (1) the synthesis and characterization of a new 1D compound through the reaction of [Nb6Cl12(CN)6]4− and [Mn(salen)]+ in MeOH–H2O; (2) solvent-mediated structural transformation of 1 into a previously reported 2D compound (1D → 2D).1
Experimental
General
(Me4N)4[Nb6Cl12(CN)6]·2MeOH and [Mn(salen)]ClO4·2H2O were synthesized according to literature methods.1,11 LiCl (99%), NbCl5 (99%, metal basis), Nb powder (99.8%, metal basis), KCN (96%) and Me4NCl (98%) were purchased from Alfa Aesar. Mn(OAc)3·2H2O (98%) and ethylenediamine (99%) were purchased from ACROS. Salicylicaldehyde was purchased from Fisher. All chemicals were used as received. Solvents EtOH, THF, MeCN and MeOH were used as received without further purification.Synthesis
Solvent-mediated structural transformation
(Me4N)Cl (100 mg, 0.9 mmol) was added to a suspension of crystals of 1 (26 mg, 0.01 mmol) in methanol (10 ml) at room temperature. The reaction mixture was left undisturbed for 2 days after which a brown microcrystalline solid formed and was separated by centrifugation, washed with water and methanol, then dried in vacuum to get 2. Anal. Calcd. for 2C46H52Cl12Mn2N12Nb6O4: C, 28.63; H, 2.72; N, 8.71%. Found: C, 28.79; H, 2.87; N, 8.23%. The phase identity and purity was determined by comparing the observed powder X-ray diffraction to that calculated from the crystal structure determined from single crystal X-ray diffraction data.Structural determination
Intensity data from a single crystal of (1) were measured at 193(2) K on a Bruker SMART APEX CCD area detector system. Data were corrected for absorption effects using the multi-scan technique (SADABS). Structure was solved and refined using the Bruker SHELXTL (Version 6.1) Software Package. The integration of the data using a triclinic cell yielded a total of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 609 reflections to a maximum θ angle of 25.00°, of which 8362 were independent (Rint = 5.05%), and 6033 (72.15%) were greater than 2σ(F2). The structure was solved and refined in the space group P
609 reflections to a maximum θ angle of 25.00°, of which 8362 were independent (Rint = 5.05%), and 6033 (72.15%) were greater than 2σ(F2). The structure was solved and refined in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No. 2), with Z = 1. All non-hydrogen atoms were refined anisotropically. O5 is fully occupied but present as coordinated water (1/3 of the time) and coordinated MeOH (2/3 of the time). Since the disordered lattice solvent could not be unambiguously modeled, the Platon Squeeze option was utilized based on the model that included the coordinated MeOH/H2O atoms only.12 Squeeze indicates 4 solvent regions in the cell corresponding to about 53 electrons/cell or approximately 2 methanol and 1.7 water molecules per cell.
(No. 2), with Z = 1. All non-hydrogen atoms were refined anisotropically. O5 is fully occupied but present as coordinated water (1/3 of the time) and coordinated MeOH (2/3 of the time). Since the disordered lattice solvent could not be unambiguously modeled, the Platon Squeeze option was utilized based on the model that included the coordinated MeOH/H2O atoms only.12 Squeeze indicates 4 solvent regions in the cell corresponding to about 53 electrons/cell or approximately 2 methanol and 1.7 water molecules per cell.
The final anisotropic full-matrix least-squares refinement on F2 with 532 variables converged to R1 = 4.70% for observed data and wR2 = 11.19% for all data. The maximum residual peak and hole on the final difference electron density map was found to be 1.152 e−/Å3 (1.25 Å from H51B) and −0.715 e−/Å3 (0.89 Å from Nb2), respectively. A summary of the most important crystal and structure refinement data is given in Table 1.
| 1 | |
|---|---|
| a *R1 = ∑(|Fo| − |Fc|)/∑|Fo|, **wR2 = {∑w[(F2o − F2c)]/∑w[(F2o)2]}0.5, w = [σ2(F2o) + (0.0490P)2 + 0.0000P] −1, where P = (F2o + 2F2c)/3]. | |
| formula | C73.33H74.07Cl12Mn4N14Nb6O13.70 |
| Mr/g | 2573.32 |
| Crystal system | Triclinic |
| Space group |
P
|
| a/Å | 12.606(3) |
| b/Å | 13.815(3) |
| c/Å | 14.013(3) |
| α/° | 78.919(2) |
| β/° | 86.321(2) |
| γ/° | 87.030(2) |
| V/Å3 | 2388.0(8) |
| Z | 1 |
| d calcd/g cm−3 | 1.789 |
| F(000) | 1272 |
| θ range° | 3.80–25.00 |
| Reflections collected/unique | 17609/8362 |
| R(int) | 0.0505 |
| R1,* wR2*, I > 2σ(I) | R1 = 0.0470, wR2 = 0.1038 |
| R1,* wR2*, all data | R1 = 0.0715, wR2 = 0.1119 |
| Goodness-of-fit on F2 | 0.972 |
| Max/mean shift in final cycle | 0.001/0.000 |
Other physical measurements
Elemental analyses were carried out by Atlantic Microlab, Inc. Thermogravimetric analyses were performed with 14 mg sample under a flow of air (40 mL/min) at a heating rate of 5 °C/min, using a Perkin-Elmer Pyris 1 TGA system. Infrared spectra were recorded as KBr pellets on a Mattson Infinity System FTIR spectrometer. The magnetic susceptibility data were collected using a SQUID (superconducting quantum interference device, MPMS, Quantum Design) magnetometer. Data were corrected for the diamagnetic moment of the sample holder, the diamagnetic contribution of the octahedral {Nb6} cluster13 and of the ligands using Pascal's constants.14X-Ray powder diffraction data were collected at room temperature using a BRUKER P4 general-purpose four-circle X-ray diffractometer equipped with a GADDS/Hi-Star detector positioned 20 cm from the sample. The goniometer was controlled using the GADDS software suite.15 The sample was mounted on tape and data were collected in transmission mode. The data ere reduced by area integration methods to produce a single powder diffraction pattern for each frame. Individual powder diffraction patterns were merged and analyzed with the program EVA to produce a single one-dimensional pattern.16Results and discussion
Crystal structure of 1
The structure of 1 is 1D coordination polymer that consists of neutral zigzag chains built of {[Mn(salen)(s)]2[Nb6Cl12(CN)6]}2− supramolecular assemblies and [Mn2(salen)2]2+ dimers linked by cyanide ligands (Fig. 1(a)). Selected bond lengths and angles are shown in Table 2. The {[Mn(salen)(s)]2[Nb6Cl12(CN)6]}2− anions are built of [Nb6Cl12(CN)6]4−cluster that connects to two [Mn(salen)(s)]+ monomers by the N end of two CN ligands that are trans to each other. The [Nb6Cl12(CN)6]4−cluster unit has an octahedral Nb6 metal core equipped with 12 edge-bridging Cl ligands and six terminal CN− ligands connected to the cluster through their carbon end. The average bond lengths and angles of Nb–Nb (2.925(2) Å), Nb–Cl (2.466(7) Å), Nb–C(2.278(9) Å), C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N (1.14(1) Å) and Nb–CN (176(1)°) are close to the corresponding values reported for other compounds containing the octahedral cyanochloride niobium clusters with 16 valence electrons available for metal-metal bonding.1,6 The IR vibration corresponding to CN stretching band is observed near 2134 cm−1 close to that found for the starting material (Me4N)4[Nb6Cl12(CN)6] (2129 cm−1).1
N (1.14(1) Å) and Nb–CN (176(1)°) are close to the corresponding values reported for other compounds containing the octahedral cyanochloride niobium clusters with 16 valence electrons available for metal-metal bonding.1,6 The IR vibration corresponding to CN stretching band is observed near 2134 cm−1 close to that found for the starting material (Me4N)4[Nb6Cl12(CN)6] (2129 cm−1).1
![Structure of compound 1. (a) The structure of {[Mn(salen)(s)]2[Nb6Cl12(CN)6]}2−heterotrimer; (b) the structure of [Mn2(salen)2]2+ dimer; (c) the neutral coordination chain; (d) the stacking of the chains viewed along the b direction. Hydrogen bonding holding the chains together is represented as dotted black lines. The salen ligand, chlorine atoms and free solvent molecules are omitted for clarity.](/image/article/2011/CE/c004119j/c004119j-f1.gif) | ||
| Fig. 1 Structure of compound 1. (a) The structure of {[Mn(salen)(s)]2[Nb6Cl12(CN)6]}2−heterotrimer; (b) the structure of [Mn2(salen)2]2+ dimer; (c) the neutral coordination chain; (d) the stacking of the chains viewed along the b direction. Hydrogen bonding holding the chains together is represented as dotted black lines. The salen ligand, chlorine atoms and free solvent molecules are omitted for clarity. | ||
| a Symmetry transformations used to generate equivalent atoms: #1 − x, −y − 1, −z + 2. b The three Nb atoms are inside an equatorial plane. c The three Nb atoms make up one face of the octahedron. d There are two kinds of [Mn(salen)]+ units. Mn(1) forms the [Mn2(salen)2]2+ dimer, and Mn(2) belongs to the heterotrimer. e O(5) is belongs to the solvent ligand. | |||
|---|---|---|---|
| [Nb6Cl12(CN)6]4− | |||
| Average Nb–Nb | 2.925(2) | Average Nb–Nb–Nbb | 90.00(4) |
| Average Nb–Cl | 2.466(7) | Average Nb–Nb–Nbc | 60.00(6) |
| Average Nb–C | 2.278(9) | Average Cl–Nb–Cl | 88.7(7) |
| Average CN |
1.14(1) | Average Nb–CN |
176(1) |
| [Mn(salen)]+ unit d | |||
| Mn(1)–O(2) | 1.894(4) | Mn(2)–O(3) | 1.864(4) |
| Mn(1)–O(1) | 1.907(4) | Mn(2)–O(4) | 1.891(4) |
| Mn(1)–N(5) | 1.985(5) | Mn(2)–N(8) | 1.978(5) |
| Mn(1)–N(4) | 1.987(5) | Mn(2)–N(7) | 1.997(5) |
| Mn(1)–N(2) | 2.206(5) | Mn(2)–N(1) | 2.258(5) |
| Mn(1)–O(1) (phenoxo bridge) | 2.405(4) | Mn(2)–O(5)#1e | 2.272(5) |
| Average CN–Mn |
158(1) | Mn(1)–O(1)–Mn(1)#1 | 102.03(16) |
In the [Mn(salen)(s)]+ monomer, each Mn(III) ion is chelated by a tetradentate salen ligand with average Mn–O = 1.88(2) Å and Mn–N = 1.98(1) Å, compared with Mn–O = 1.882 (1) Å and Mn–N = 1.983(1) Å in [Mn(salen)Cl(H2O)]).17 The distorted octahedral coordination environment of Mn(III) is completed by the coordination of one cyanide ligand with Mn–NCN = 2.206(5) Å and Mn–NC = 158(1)°, and one solvent molecule with Mn–O = 2.272(5) Å.
The [Mn2(salen)2]2+ dimers are formed through phenoxo bonding between two Mn(salen) complexes with Mn⋯Mn = 3.366(1) Å, Mn–O = 2.405(4) Å and ∠Mn–O–Mn = 102.1(2)°, within the range reported for other Mn(salen)-type dimers (3.19–4.38 Å and 91.1–102.4° respectively).18 Each dimer uses its remaining two coordination sites to link to two cyano ligands from the cluster with Mn–NCN distance of 2.206(5) Å and CN–Mn angle of 159.2(5)°.
The neutral chains are stacked parallel to each other along the crystallographic [1 0 −1] axis. Hydrogen bonds between the remaining non-bridging cyanides and the coordinated solvent molecules (O5–H51⋯N3 = 2.742(7) Å, symmetry code: x, y + 1, z) connect the chains into layers that stack perpendicular to the c direction (Fig. 1(d)). Adjacent layers are related to each other by an inversion center and are separated by disordered solvent molecules.
Solvent-mediated structural transformation
When samples of 1 consisting of large crystals were suspended in a methanol solution containing (Me4N)Cl, an interesting structural transformation took place in which 1 was transformed into compound 2 which has a 2D anionic 4,4′-network built of [Nb6Cl12(CN)6]4− and [Mn(salen)]+ nodes linked by CN ligands.1 The layers in 2 stack on top of each other in a staggered fashion and generate channels where (Me4N)+ are located. The transformation is complete as indicated by PXRD (Fig. 2), elemental analysis and IR. However, 2 was found to remain stable for weeks when suspended in neat methanol or in a methanolic solution containing [Mn(salen)]+, which indicates that the transformation is irreversible.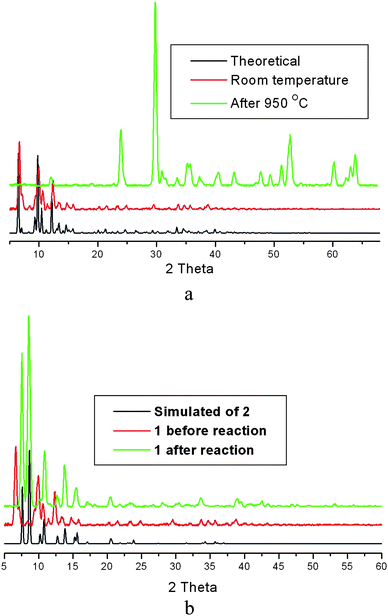 | ||
| Fig. 2 (a) Powder X-ray diffraction patterns of 1: pattern simulated from crystal structure in black; observed pattern at room temperature in red; observed pattern after being heated to 950 °C in green. (b) A comparison of the powder patterns of 2 (simulated), 1 and after the reaction of 1 with (Me4N)Cl. | ||
The fact that large and well shaped crystals of compound 1 are transformed into microcrystalline powder of 2 and that the transformation is accompanied by siginficant structural changes, seems to indicate that the transformation is solvent-mediated and cation-induced. The mechanism of such transformation can be described as follows (Scheme 1): the 1D framework of 1 slowly dissociates to form the dication [Mn2(salen)2]2+ and the anionic [Mn(salen)2(Nb6Cl12(CN)6)]2−heterotrimer built of [Nb6Cl12(CN)6]4− linked to two [Mn(salen)]+ complexes through two transcyanide ligands. In a subsequent step the heterotrimer assembles in the presence of (Me4N)+ cations to form the 2D 4,4′-net in which the layers pack in a staggered way. As 2 slowly crystallizes (precipitates), the solubility equilibrium is shifted toward the product and more of 1 is dissolved to form more 2 until complete transformation of 1. Similar solvent-mediated processes have been observed in the structural transformations of cluster-based 2D frameworks6 and anion exchange in Ag-based coordination polymers.10
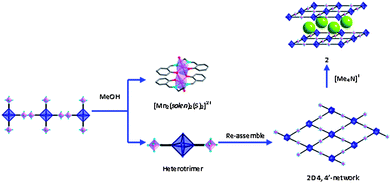 | ||
| Scheme 1 Possible mechanism for the solvent-mediated structural transformation of 1. | ||
Thermal stability
The thermal stability of 1 was studied by thermal analysis under air flow using polycrystalline samples. The result showed that the compound gradually decomposes upon heating from RT to 700 °C. PXRD showed that a mixture of MnNb2O6 and Mn3O4 (% weight loss (obs.) = 42.31%; (calc.) = 42.22%) is formed after 700 °C (Fig. S1, ESI†).19Magnetic properties
The magnetic susceptibility of 1 was measured between 300 and 5 K on microcrystalline powders. Fig. 3 shows the data in the forms of μeffvs T and 1/χMvs. T. At room temperature the μeff values for 1 is 4.90 μB, compared with the spin-only value (4.90 μB) for four MnIII ions with g = 2.0 and S = 2. As the temperature decreases μeff slowly decreases until ca. 70 K, then sharply decreases to reach 3.67 μB at 5 K, indicating weak antiferromagnetic coupling between MnIII ions. The χm−1vs. T plot follows Curie–Weiss law with C = 0.62 emu mol−1 K−1 and θ = −4.91 K also indicating antiferromagnetic coupling, which can be attributed to intradimer coupling. The magnetic properties were modeled using the Heisenberg dimer model with the Hamiltonian Ĥ = −2JŜ1Ŝ2 giving the van Vleck equation:
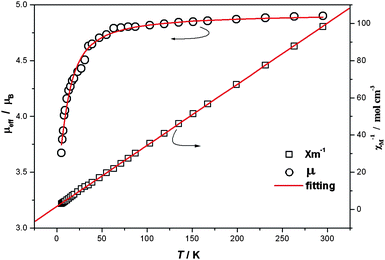 | ||
| Fig. 3 Temperature dependence of magnetic susceptibility of 1 (5–300 K at 1000 G). | ||
The best fit gave: g = 2.011(5), J = −1.50(5) cm−1 as shown in Fig. 3. The result is comparable with that observed in compounds containing Mn(salen)-type dimers as building blocks.18
Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. DMR-0446763. We are grateful to Dr Gordon Yee from the chemistry Department at Virginia Tech for magnetic susceptibility data collection.Notes and references
- H. J. Zhou, C. S. Day and A. Lachgar, Chem. Mater., 2004, 16, 4870 CrossRef CAS.
- F. A. Cotton and R. A. Walton, Multiple bonds between metal atoms, second ed., Oxford University Press, 1993 Search PubMed; E. J. Welch and J. R. Long, Prog. Inorg. Chem., 2005, 54, 1 Search PubMed; J.-C. P. Gabriel, K. Boubekeur, S. Uriel and P. Batail, Chem. Rev., 2001, 101, 2037 CAS; H. D. Selby, B. K. Roland and Z. P. Zheng, Acc. Chem. Res., 2003, 36, 933 CrossRef CAS; H. J. Zhou and A. Lachgar, Eur. J. Inorg. Chem., 2007, 1053 CrossRef CAS.
- E. G. Tulsky, N. R. M. Crawford, S. A. Baudron, P. Batail and J. R. Long, J. Am. Chem. Soc., 2003, 125, 15543 CrossRef CAS; D. H. Johnston, C. L. Stern and D. F. Shriver, Inorg. Chem., 1993, 32, 5170 CrossRef CAS; Y. V. Mironov, V. E. Fedorov, I. Ijjaali and J. A. Ibers, Inorg. Chem., 2001, 40, 6320 CrossRef CAS.
- N. G. Naumov, A. V. Virovets, M. N. Sokolov, S. B. Artemkina and V. E. Federov, Angew. Chem., Int. Ed., 1998, 37, 1943 CrossRef CAS; M. V. Bennett, M. P. Shores, L. G. Beauvais and J. R. Long, J. Am. Chem. Soc., 2000, 122, 6664 CrossRef CAS; Y. Kim, S. M. Park, W. Nam and S. J. Kim, Chem. Commun., 2001, 1470 RSC.
- B. B. Yan, H. J. Zhou and A. Lachgar, Inorg. Chem., 2003, 42, 8818 CrossRef CAS; N. G. Naumov, D. V. Soldatov, J. A. Ripmeester, S. B. Artemkina and V. E. Federov, Chem. Commun., 2001, 571 RSC.
- H. J. Zhou, C. S. Day and A. Lachgar, Cryst. Growth Des., 2006, 6, 2384 CrossRef CAS; H. J. Zhou, K. C. Strates, M. A. Munoz, K. J. Little, D. M. Pajerowski, M. W. Meisel, T. A. Talham and A. Lachgar, Chem. Mater., 2007, 19, 2238 CrossRef CAS; J. J. Zhang and A. Lachgar, J. Am. Chem. Soc., 2007, 129, 250 CrossRef CAS; J. J. Zhang, H. J. Zhou and A. Lachgar, Angew. Chem., Int. Ed., 2007, 46, 4995 CrossRef CAS; J. J. Zhang, Y. Zhao, S. A. Gamboa and A. Lachgar, Cryst. Growth Des., 2008, 8, 172 CrossRef CAS; J. J. Zhang, Y. Zhao, S. A. Gamboa, M. Muñoz and A. Lachgar, Eur. J. Inorg. Chem., 2008, 2982 CrossRef CAS.
- R. Robson in Comprehensive Supramolecular Chemistry, Pergamon, New York, 1996, ch22, p733 Search PubMed; M. Eddaoudi, B. Moler, H. Li, B. Chen, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 Search PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS; S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109 RSC.
- D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior and M. J. Rosseinsky, Acc. Chem. Res., 2005, 38, 273 CrossRef CAS.
- A. N. Khlobystov, N. R. Champness, C. J. Roberts, S. J. B. Tendler, C. Thompson and M. Schröder, CrystEngComm, 2002, 4, 426 RSC; R. Custelcean and B. A. Moyer, Eur. J. Inorg. Chem., 2007, 1321 CrossRef CAS.
- H. Li, J. Z. Zhong, C. Y. Duan, X. Z. You, T. C. W. Mak and B. Wu, J. Coord. Chem., 1997, 41, 183 CrossRef CAS.
- SQUEEZE (handling of disordered solvents in structure refinement) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 Search PubMed.
- J. G. Converse and R. E. McCarley, Inorg. Chem., 1970, 9, 1361 CrossRef CAS.
- O. Kahn, Molecular Magnetism, Wiley-VCH, New York, 1993, P.3 Search PubMed.
- GADDS V4.1.14 “General Area Detector Diffraction System Program for Instrument Control and zData Collection” BRUKER AXS Inc., 5465 East Cheryl Parkway, Madison, WI 53711–5373 USA Search PubMed.
- EVA V8.0 “Graphics Program for 2-dimensional Data evaluation and Presentation” BRUKER AXS Inc., 5465 East Cheryl Parkway, Madison, WI 53711–5373 USA Search PubMed.
- A. Panja, N. Shaikh, M. Ali, P. Vojtisek and P. Banerjee, Polyhedron, 2003, 22, 1191 CrossRef CAS.
- S. Saha, D. Mal, S. Koner, A. Bhattacherjee, P. Gutlich, S. Mondal, M. Mukherjee and K.-I. Okamoto, Polyhedron, 2004, 23, 1811 CrossRef CAS; H. Miyasaka, R. Clérac, T. Ishii, H. C. Chang, S. Kitagawa and M. Yamashita, J. Chem. Soc., Dalton Trans., 2002, 1528 RSC and references therein Z. L. Lu, M. Yuan, F. Pan, S. Gao, D. Q. Zhang and D. B. Zhu, Inorg. Chem., 2006, 45, 3538 Search PubMed.
- H. Weitzel, Z. Kristallogr., 1976, 144, 238 (ICSD code for MnNb2O6: 31944) L. M. Kuznetsov, A. N. Tsvigunov and K. P. Burdina, Geokhimiya, 1979, 254 Search PubMed (ICSD code for Mn3O4: 30005).
Footnote |
| † Electronic supplementary information (ESI) available: Plots of TGA and XRD of the phase after being heated to 950 °C. CCDC reference number 762244. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c004119j |
| This journal is © The Royal Society of Chemistry 2011 |