DOI:
10.1039/C000586J
(Paper)
CrystEngComm, 2011,
13, 230-242
Diverse mixed-ligand metal complexes with in situ generated 5-(pyrazinyl)tetrazolate chelating-bridging ligand: in situ synthesis, crystal structures, magnetic and luminescent properties†
Received
13th January 2010
, Accepted 11th June 2010
First published on 3rd September 2010
Abstract
The purpose of this work is to systematically investigate the influence of the functional co-ligands and metal ions on the self-assembly and properties of the in situ generated 5-(pyrazinyl)tetrazolate (ptz−)-based metal complexes. A series of nine metal complexes, {[Co3(H2O)4(ptz)2(btec)]·5H2O}n (1), [Cu2(H2O)4(ptz)2(H2btec)]n (2), [Hg2(ptz)2Cl2]n (3), [Hg3(H2O)2(ptz)4(μ1,1-N3)2]·2H2O (4), Hg(H2O)2(ptz)2 (5), [Cd2(H2O)2(ptz)2(ox)]n (6), [Cd(H2O)(ptz)I]n (7), {[Cd(H2O)(ptz)X]·H2O}n (X = Br for 8 and Cl for 9) (btec = 1,2,4,5–benzenetetracarboxylate and ox = oxalate), was obtained by a subtle variation of co-ligands and metal ions, in which the HgII ion is firstly observed as an efficient Lewis acid to catalyze the in situ reaction. Significantly, resulted from the cooperative and/or competitive binding between the mixed ligands and the metal ions with different coordination geometry, complexes 1–9 feature interesting structural subunits (mono-, bi- and tri-metallic core) and various dimensionalities ranged from polymeric three-dimensional (3D) frameworks to discrete zero-dimensional (0D) entities. Three high-dimensional complexes with a binodal (3,8)-connected tfz-d microporous framework for 1, a trinodal (3,4)-connected unusual (6·82)2(6·83·102) topology for 3, and a six-connected α-polonium-type network for 6 are observed, respectively. Complexes 2 and 7–9 are bi-metallic node-based 2D layers (7) and 1D chains (2, 8, and 9). In contrast, the rarely linear tri-metallic core and commonly observed mononuclear entities are generated for 4 and 5, respectively. Additionally, CuII-based polymer 2 displays weak intra-dimeric antiferromagnetic coupling interaction. Complexes 1 and 3–9 with considerable metal ion-dependent thermal stability exhibit ptz−-based photoluminescence with variable intensity.
Introduction
Self-assembled frameworks containing metal ions and mixed organic bridging ligands are of great current interest due to their unexpected coordination architectures and unique functions.1 Up to now, the exactly structural predictions for the complexes bearing polydentate N- and O-containing ligands and metal ions with variable coordination geometry are extremely difficult and challenging, because the overall structures of the target complexes highly depend on the nature of the metal ions and/or the ligands, the synthetic conditions as well as the preparation methods. In this regard, polydentate 5-substituted-1H-tetrazolate-based ligands generated by in situ reactions, such as pyrazinyl-,2–5 pyrimidyl-,6–7 position isomeric pyridyl-,8–10 phenyl-,11–13 amino-,14 have been extensively employed as excellent chelating and/or bridging linkers to construct novel metal complexes with interesting optical properties, due to their diverse connectivity modes and high structural stability. For example, a series of metal-directed (ZnII,2, 4 CdII,2 CuII,2–3 AgI,2 CoII,3 and MnII 4) binary supramolecular architectures bearing the in situ generated 5-(pyrazinyl)tetrazolate (ptz−) ligand have been recently reported, indicating that the different metal ions upon the self assembly process help to build diverse coordination geometry, while the hydrogen bonding and other supramolecular interactions are responsible for the ordered extension of the architecture dimensionality. More recently, Bu and co-workers15 have explored the assembly behaviour of group IIB metal ions with the pre-synthesized ptz ligand as well as the emission properties of the resulting complexes, finding the luminescence and structural dependence on the metal ions and preparing conditions. Herein, as the continuing investigations on the influence of the co-ligand and metal ions on the structures and properties of polyazolate-based metal complexes,16–21 we report the synthesis and optical/magnetic properties of a series of nine co-ligand-assisted ternary ptz−-based metal complexes. It is well known that the presence of functional co-ligand can significantly produce multiple effects on the core ligand in addition to the metal coordination sphere, which ultimately govern the overall structures of the mixed-ligand complexes. As a result, a series of nine co-ligand-regulated ternary ptz−-based metal complexes with interesting subunits (mono-, bi- and tri-metallic core) and various dimensions (0D, 1D, 2D and 3D) was generated, which includes three high-dimensional frameworks with a (3,8)-connected tfz-d net for 1, a trinodal (3,4)-connected unusual (6·82)2(6·83·102) topology for 3, and a six-connected α-polonium-type network for 6, one bimetallic node-based 2D layer (7), three bimetallic node-based 1D chains (2, 8, and 9), one unusual linear trimetallic core for 4, and commonly observed mononuclear entities for 5, respectively. Undoubtedly, such interesting structural diversity is significantly due to the spontaneous combinations of the variable coordination modes between the core ptz− ligand and the specific co-ligands with either bridging or terminal function. Besides, we observed for the first time that HgII ion can be used as effective Lewis acid catalyst to construct the in situ-generated coordination framework. Thus, the Lewis acid catalysts available in in situ reaction cover ZnII, CuI/II, AgI, CoII, CdII, NiII, MnII, FeII and HgII salts. On the other hand, resulted from the nature of the metal ion and ptz− ligands, the resulting complexes display favorable emissions for 1 and 3–9 and weak antiferromagnetic coupling interaction for 2.
Experimental
Materials and methods
All reagents were commercially purchased (2-cyanopyrazine was from TCI and other analytical-grade reagents were from Tianjin Chemical Reagent Factory) and used without further purification. Doubly deionized water was used for the conventional synthesis. Elemental analyses for C, H and N were carried out with a CE-440 (Leeman Labs) analyzer. Fourier transform (FT) IR spectra (KBr pellets) were taken on an Avatar-370 (Nicolet) spectrometer in the range of 4000–400 cm−1. Thermogravimetric analysis (TGA) experiments were carried out on Shimadzu simultaneous DTG-60A compositional analysis instrument from room temperature to 800 °C under N2 atmosphere at a heating rate of 5 °C min−1. Fluorescence spectra of the polycrystalline powder samples were performed on a Cary Eclipse fluorescence spectrophotometer (Varian) equipped with a xenon lamp and quartz carrier at room temperature. Variable-temperature susceptibility measurement of 2 was carried out at an applied DC field of 1000 Oe from 4 to 300 K temperature range on a Quantum Design (SQUID) magnetometer MPMS-XL-7.
Preparation of complexes 1–9
{[Co3(H2O)4(ptz)2(btec)]·5H2O}n (1).
CoCl2·6H2O (47.6 mg, 0.2 mmol), H4btec (25.4 mg, 0.1 mmol), 2-cyanopyrazine (10.5 mg, 0.1 mmol), NaN3 (13.0 mg, 0.2 mmol) and water (10 mL) were sealed in a parr Teflon-lined stainless steel vessel (23 mL) and heated to 160 °C for 96 h under autogenous pressure. Orange block-shaped crystals suitable for X-ray analysis were obtained directly, washed with ethanol, and dried in air. Yield: 72% based on 2-cyanopyrazine. Anal. calcd for C20H26Co3N12O17: C 27.20, H 2.97, N 19.03%. Found: C 27.14, H 3.02, N 19.15%. IR (KBr, ν/cm−1): 3406 (br), 3102 (w), 1600 (s), 1572 (vs), 1485 (m), 1415 (s), 1370 (vs), 1318 (w), 1280 (w), 1170 (w), 1149 (w), 1074 (w), 1045 (m), 938 (w), 870 (m), 810 (w), 770 (w), 580 (m), 541 (m), 433 (m).
[Cu2(H2O)4(ptz)2(H2btec)]n (2).
Hydrothermal reaction of CuCl2·2H2O (34.1 mg, 0.2 mmol), H4btec (25.4 mg, 0.1 mmol), 2-cyanopyrazine (10.5 mg, 0.1 mmol), NaN3 (13.0 mg, 0.2 mmol) and water (10 mL) at 100 °C for two days generated blue block-shaped crystals of 2. Yield: 62% based on 2-cyanopyrazine. Anal. calcd for C10H9N6CuO6: C 32.22, H 2.43, N 22.55%. Found: C 32.45, H 2.24, N 22.43%. IR (KBr, ν/cm−1): 3425 (br), 3097 (w), 1712 (ms), 1631 (s), 1564 (s), 1502 (w), 1425 (s), 1382 (ms), 1346 (w), 1295 (w), 1222 (ms), 1171 (m), 1116 (ms), 1073 (m), 1046 (m), 945 (w), 862 (w), 829 (w), 592 (w), 530 (w), 445 (w).
[Hg2(ptz)2Cl2]n (3).
To an aqueous solution (6 mL) containing NaN3 (13.0 mg, 0.2 mmol) and HgCl2·0.5H2O (54.3 mg, 0.2 mmol) was slowly added an ethanol solution (6 mL) of 2-cyanopyrazine (10.5 mg, 0.1 mmol) and H4btec (12.7 mg, 0.05 mmol) with constant stirring. After further stirring for one hour, the resulting mixture was filtered. Colourless strip-shaped crystals suitable for X-ray analysis were obtained by evaporation of the filtrate in one week, washed with ethanol, and dried in air. Yield: 43% based on 2-cyanopyrazine. Anal. calcd for C10H6Cl2Hg2N12: C 15.67, H 0.79, N 21.93%. Found: C 15.70, H 0.84, N 21.77%. IR (KBr, ν/cm−1): 3073 (w), 1421 (s), 1400 (ms), 1375 (w), 1294 (w), 1170 (ms), 1116 (w), 1095 (w), 1063 (w), 1025 (s), 862 (m), 762 (m), 577 (w), 514 (w).
[Hg3(H2O)2(ptz)4(μ1,1-N3)2]·2H2O (4).
A methanol solution (6 mL) containing 2-cyanopyrazine (10.5 mg, 0.1 mmol), H4btec (12.7 mg, 0.05 mmol) and NaN3 (13.0 mg, 0.2 mmol) was carefully layered onto a buffer layer of ethyl acetate (2 mL) in a straight glass tube, below which an aqueous solution of Hg(NO3)2·H2O (33.0 mg, 0.1 mmol) was placed. Upon slow evaporation of the mixture at room temperature, colourless block-shaped crystals suitable for X-ray analysis were obtained on the wall of the tube within two weeks, washed with ethanol and water and dried in air. Yield: 26% based on 2-cyanopyrazine. Anal. calcd for C10H10Hg1.5N15O2: C 17.84, H 1.50, N 31.21%. Found: C 17.70, H 1.74, N 31.07%. IR (KBr, ν/cm−1): 3457 (br), 3063 (w), 2040 (s), 1624 (ms), 1529 (w), 1448 (w), 1398 (m), 1357 (w), 1169 (w), 1136 (m), 1057 (w), 1021 (m), 861 (w), 764 (w), 637 (w), 522 (w), 460 (w).
Hg(H2O)2(ptz)2 (5).
Complex 5 was prepared by adopting the same synthetic procedures as 3 only increasing the amount of NaN3 to 0.3 mmol (19.5 mg). Colourless block-shaped crystals suitable for X-ray analysis were obtained by evaporation of the filtrate in one week, washed with ethanol, and dried in air. Yield: 22% based on 2-cyanopyrazine. Anal. calcd for C5H5Hg0.5N6O: C 22.63, H 1.90, N 31.66%. Found: C 22.41, H 1.87, N 31.75%. IR (KBr, ν/cm−1): 3427 (br), 3059 (w), 1656 (w), 1562 (w), 1528 (w), 1398 (vs), 1356 (w), 1171 (m), 1138 (s), 1057 (ms), 1022 (s), 864 (m), 766 (w), 522 (w).
[Cd2(H2O)2(ptz)2(ox)]n (6).
Complex 6 was prepared by the same procedures as 1 except that CoCl2·6H2O and H4btec were replaced by CdCl2·2.5H2O (45.7 mg, 0.2 mmol) and H2ox·2H2O (12.7 mg, 0.1 mmol), respectively. Colourless block-shaped crystals suitable for X-ray analysis were obtained directly, washed with ethanol, and dried in air. Yield: 64% based on 2-cyanopyrazine. Anal. calcd for C6H5CdN6O3: C 22.41, H 1.57, N 26.14%. Found: C 22.45, H 1.49, N 26.17%. IR (KBr, ν/cm−1): 3322 (br), 3105 (w), 1619 (vs), 1465 (w), 1415 (s), 1377 (m), 1308 (s), 1171 (w), 1154 (m), 1133 (w), 1075 (w), 1047 (m), 1027 (w), 858 (w), 784 (m), 757 (w), 698 (w), 635 (w), 521 (m), 480 (w), 438 (w).
[Cd(H2O)(ptz)I]n (7).
CdI2·4H2O (73.2 mg, 0.2 mmol), 2-cyanopyrazine (10.5 mg, 0.1 mmol), NaN3 (26.0 mg, 0.4 mmol), and water (10 mL) were sealed in a parr Teflon-lined stainless steel vessel (23 mL) and heated to 160 °C for 72 h under autogenous pressure. Pale-yellow block-shaped crystals suitable for X-ray analysis were obtained directly, washed with ethanol, and dried in air. Yield: 39% based on 2-cyanopyrazine. Anal. calcd for C5H5CdIN6O: C 14.85, H 1.25, N 20.78%. Found: C 14.74, H 1.30, N 20.41%. IR (KBr, ν/cm−1): 3465 (br), 3087 (w), 1641 (m), 1536 (w), 1459 (w), 1408 (s), 1376 (m), 1287 (w), 1233 (w), 1165 (ms), 1073 (ms), 1041 (s), 852 (w), 762 (m), 603 (m), 523 (m), 417 (m).
{[Cd(H2O)(ptz)Br]·H2O}n (8).
Complex 8 was prepared by adopting the same procedures as 7 only using CdBr2·4H2O (68.8 mg, 0.2 mmol) instead of CdI2·4H2O. Colourless block-shaped crystals suitable for X-ray analysis were obtained directly, washed with ethanol, and dried in air. Yield: 47% based on 2-cyanopyrazine. Anal. calcd for C5H7BrCdN6O2: C 16.00, H 1.88, N 22.38%. Found: C 16.16, H 1.90, N 22.42%. IR (KBr, ν/cm−1): 3201 (br), 3076 (w), 2074 (s), 1623 (m), 1559 (m), 1408 (s), 1375 (w), 1342 (w), 1280 (w), 1167 (m), 1147 (ms), 1070 (m), 1039 (ms), 867 (w), 761 (w), 663 (w), 510 (w), 479 (w), 422 (w).
{[Cd(H2O)(ptz)Cl]·H2O}n (9).
CdCl2·2.5H2O (45.7 mg, 0.2 mmol), 2-cyanopyrazine (10.5 mg, 0.1 mmol), NaN3 (26.0 mg, 0.4 mmol) and water (10 mL) were sealed in a parr Teflon-lined stainless steel vessel (23 mL) and heated to 160 °C for 72 h under autogenous pressure. After cooling to room temperature, the mixture was filtered. Colourless block-shaped crystals suitable for X-ray analysis were obtained by evaporation of the filtrate within two weeks, washed with ethanol, and dried in air. Yield: 51% based on 2-cyanopyrazine. Anal. calcd for C5H7CdClN6O2: C 18.14, H 2.13, N 25.39%. Found: C 18.16, H 2.08, N 25.42%. IR (KBr, ν/cm−1): 3218 (br), 3081 (w), 2076 (s), 1626 (m), 1559 (m), 1409 (s), 1376 (w), 1341 (w), 1282 (w), 1168 (m), 1147 (ms), 1071 (m), 1039 (ms), 869 (w), 760 (w), 668 (w), 510 (w), 479 (w), 422 (w).
Diffraction intensities for complexes 1–9 were collected using a Bruker APEX–II CCD diffractometer equipped with graphite-monochromated Mo-Kα radiation with radiation wavelength 0.71073 Å by using the φ–ω scan technique at 296(2) K. There was no evidence of crystal decay during data collection. Semiempirical absorption corrections were applied (SADABS), and the program SAINT was used for integration of the diffraction profiles.22 The structures were solved by direct methods and refined with the full–matrix least–squares technique using the SHELXS-97 and SHELXL-97 programs.23 Anisotropic thermal parameters were assigned to all non-hydrogen atoms. Split occupancy of 0.5 was applied to the disordered lattice water molecule O(9) of 1. And one coordinated water molecule (O6) in 2 was found disordered over two positions and refined with the sum of the occupation factors restrained to one (0.765 for O(6) and 0.235 for O(6′), respectively). The organic hydrogen atoms were generated geometrically. The H atoms of the water molecules except for the splitting water were located from difference maps and refined with isotropic temperature factors. The crystallographic data and selected bond lengths and angles for 1–9 were listed in Tables 1–5, respectively.
Table 1 Crystal and structure refinement data for 1–9a
| |
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
|
R
1 = Σ‖Fo| − |Fc‖/|Fo|.
wR2 = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2.
|
| Empirical formula |
C20H26Co3N12O17 |
C10H9CuN6O6 |
C10H6Cl2Hg2N12 |
C10H10Hg1.5N15O2 |
C5H5Hg0.5N6O |
C6H5CdN6O3 |
C5H5CdIN6O |
C5H7BrCdN6O2 |
C5H7CdClN6O2 |
| FW/g mol−1 |
883.32 |
372.77 |
766.35 |
673.22 |
265.45 |
321.56 |
404.45 |
375.48 |
331.02 |
| Crystal system |
Triclinic |
Triclinic |
Orthorhombic |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
Pbca
|
P21/c |
P21/c |
P21/n |
P21/c |
P21/n |
P21/c |
| Crystal size/mm |
0.24 × 0.23 × 0.22 |
0.18 × 0.14 × 0.13 |
0.20 × 0.16 × 0.15 |
0.22 × 0.20 × 0.18 |
0.20 × 0.18 × 0.15 |
0.26 × 0.23 × 0.22 |
0.22 × 0.20 × 0.17 |
0.22 × 0.20 × 0.18 |
0.23 × 0.22 × 0.20 |
|
a/Å |
8.2152(4) |
7.8365(10) |
16.1407(6) |
7.2819(6) |
5.5940(9) |
5.8768(5) |
7.276(2) |
7.4195(11) |
7.3629(3) |
|
b/Å |
8.9387(4) |
8.2326(11) |
6.9435(2) |
17.4904(16) |
12.643(2) |
13.1226(12) |
10.159(3) |
13.9422(19) |
13.9028(6) |
|
c/Å |
12.3961(5) |
10.7944(15) |
30.3299(11) |
14.8161(10) |
11.2431(16) |
11.5664(11) |
16.055(5) |
9.9089(14) |
11.8372(4) |
|
α/° |
109.8460(10) |
111.154(2) |
90.00 |
90.00 |
90 |
90 |
90 |
90 |
90 |
|
β/° |
92.6510(10) |
95.728(2) |
90.00 |
115.224(3) |
106.662(7) |
94.7880(10) |
112.545(11) |
95.093(2) |
123.425(2) |
|
γ/° |
106.7250(10) |
94.674(2) |
90.00 |
90.00 |
90 |
90 |
90 |
90 |
90 |
|
V/Å3 |
809.46(6) |
641.03(15) |
3399.2(2) |
1707.1(2) |
761.8(2) |
888.88(14) |
1096.0(6) |
1021.0(3) |
1011.31(7) |
|
Z
|
1 |
2 |
8 |
4 |
4 |
4 |
4 |
4 |
4 |
|
θ range/° |
1.77–25.01 |
2.04 – 25.00 |
1.34–25.01 |
1.91–25.01 |
2.48–25.01 |
2.35–25.00 |
2.43–25.00 |
2.92–25.01 |
3.14–25.00 |
|
μ/mm−1 |
1.614 |
1.751 |
18.383 |
13.539 |
10.140 |
2.460 |
4.788 |
6.043 |
2.414 |
|
F(000) |
447 |
376 |
2752 |
1244 |
500 |
620 |
744 |
712 |
640 |
| Limiting indices |
−9 ≤ h ≤ 9 |
−6 ≤ h ≤ 9 |
−19 ≤ h ≤ 10 |
−8 ≤ h ≤ 8 |
−6 ≤ h ≤ 6 |
−6 ≤ h ≤ 6 |
8 ≤ h ≤ 8 |
−8 ≤ h ≤ 8 |
−8 ≤ h ≤ 8 |
| −10 ≤ k ≤ 10 |
−9 ≤ k ≤ 9 |
−8 ≤ k ≤ 7 |
−20 ≤ k ≤ 20 |
−15 ≤ k ≤ 13 |
−8 ≤ k ≤ 15 |
12 ≤ k ≤ 6 |
−13 ≤ k ≤ 16 |
−16 ≤ k ≤ 12 |
| −14 ≤ l ≤ 1 |
−12 ≤ l ≤ 12 |
−31 ≤ l ≤ 36 |
−17 ≤ l ≤ 13 |
−13 ≤ l ≤ 9 |
−13 ≤ l ≤ 13 |
19 ≤ l ≤ 19 |
−11 ≤ l ≤ 9 |
−11 ≤ l ≤ 14 |
| Data/restraints/parameters |
2823/0/241 |
2235/0/213 |
2958/0/235 |
2965/0/259 |
1333/0/115 |
1556/0/145 |
1935/0/128 |
1760/0/136 |
1757/0/136 |
| GOF |
1.023 |
1.036 |
1.050 |
1.060 |
1.052 |
1.063 |
1.053 |
1.055 |
1.076 |
|
R
int
|
0.0107 |
0.0111 |
0.0552 |
0.0554 |
0.0340 |
0.0142 |
0.0268 |
0.0163 |
0.0213 |
|
R
1
a, wR2b [I > 2σ(I)] |
0.0280/0.0725 |
0.0305/0.0722 |
0.0336/0.0859 |
0.0282/0.0710 |
0.0326/0.0870 |
0.01570.0396 |
0.02020.0525 |
0.04110.1052 |
0.03550.0775 |
|
R
1, wR2 [all data] |
0.0310/0.0743 |
0.0390/0.0774 |
0.0373/0.0881 |
0.0327/0.0734 |
0.0389/0.0931 |
0.0164/0.0400 |
0.0221/0.0533 |
0.0430/0.1063 |
0.0361/0.0778 |
| Residuals/e Å−3 |
0.447 and −0.534 |
0.447 and −0.497 |
1.995 and −1.740 |
1.114 and −1.840 |
0.712 and −1.999 |
0.264 and −0.525 |
0.603 and −0.440 |
1.112 and −1.292 |
0.926 and −0.778 |
Results and discussion
In order to explore the influence of co-ligands and metal ions on the in situ generated metal complexes, in situ reactions of 2-cyanopyrazine and sodium azide in the presence of Cu2+, Co2+, Ni2+,24 Hg2+, Cd2+ and aromatic/aliphatic acid (H4btec and H2ox) as well as halide anions (Cl−, Br−, and I−) were performed under suitable reaction conditions (see Scheme 1) and preparation methods (hydrothermal, evaporation and diffusion). As a result, complexes 1 and 2 with the same mixed ligands but different metal centers were obtained by changing the hydrothermal temperature (160 °C vs. 100 °C). Unexpectedly, the different reaction temperature essentially led to two different deprotonated forms of the H4btec co-ligand in the two complexes. The three HgII-ptz−-based complexes, 3–5, were firstly generated by in situ reactions under mild reaction conditions.15 Thus, the effective Lewis acid catalysts in the in situ reaction are extensively extended from ZnII to CdII, MnII, CuI/II, NiII, CoII, AgI, FeII and HgII ions. Notably, the stoichiometric H4btec species in preparation of 3–5 prefers to adjust the pH value of the medium, rather than acting as a co-ligand. Additionally, the molar ratio of NaN3 to HgII salts was observed to significantly tune the final structure of the complexes. For example, high-dimensional framework of 3 can be easily transformed into the discrete mononuclear 5 by increasing the molar ratio from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (NaN3:HgII) to 1.5:1. And upon further increment of the molar ratio up to 2:1, complex 4 with unusual trinuclear motif can be obtained, although the preparation method of the three complexes is slightly different from each other (evaporation for 3 and 5vs. diffusion for 4). On the other hand, hydrothermal reactions at 160 °C afforded four CdII-based mixed-ligand complexes with short aliphatic carboxylate for 6 and comparable halide anions (I−, Br− and Cl−) for 7–9, respectively. Therefore, the suitable temperature in the ptz−−based in situ reaction plays much more important than other possible factors that influenced the formation of the target complexes.
:1 (NaN3:HgII) to 1.5:1. And upon further increment of the molar ratio up to 2:1, complex 4 with unusual trinuclear motif can be obtained, although the preparation method of the three complexes is slightly different from each other (evaporation for 3 and 5vs. diffusion for 4). On the other hand, hydrothermal reactions at 160 °C afforded four CdII-based mixed-ligand complexes with short aliphatic carboxylate for 6 and comparable halide anions (I−, Br− and Cl−) for 7–9, respectively. Therefore, the suitable temperature in the ptz−−based in situ reaction plays much more important than other possible factors that influenced the formation of the target complexes.
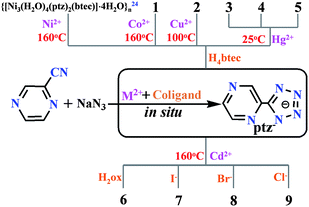 |
| | Scheme 1 Preparation details for complexes 1–9. | |
In the FT-IR spectra, the strong bands appeared above 3200 cm−1 for all the complexes but 3 should be ascribed to the stretching vibrations of O–H, suggesting the presence of free and/or coordinated water molecule.25 The characteristic bands for weak aromatic C–H vibrations were located at ca. 3100 cm−1 and the skeletal vibrations of the aromatic ring were observed in the 1400–1626 cm−1 region for 1–9. Additionally, the absence of the bands at 2200 cm−1 for stretching vibrations of cyano group, together with the new bands emerged at 1400–1500 cm−1 for the stretching vibrations of a tetrazole group, jointly suggested the formation of the ptz− ligand.2 For the three complexes with the organic acid co-ligand, the absence of characteristic band at ca. 1700 cm−1 in both 1 and 6 indicates the complete deprotonation of the carboxylic acid; while the appearance of the peak at 1712 cm−1 in 2 suggested the existence of protonated –COOH. Correspondingly, the asymmetric (νas) and symmetric vibrations (νs) of the carboxylate group were observed at 1600, 1572 and 1415, 1370 cm−1 for 1, 1564, 1631, 1564 and 1425, 1382 cm−1for 2, 1619 and 1415 cm−1 for 6, respectively. Their differences (Δv) between vas(COO−) and vs(COO−) suggested the potentially variable binding modes of the carboxylate groups.25 For 4, the strong band at 2040 cm−1 for the asymmetric stretching of N3−, together with the medium bands locating at 1169 and 1357 cm−1for the vs(N3−) stretching confirmed the existence of the end-to-on bridging mode of azide anion.26–28 Thus, the IR results were in agreement with the single-crystal X-ray diffractions.
Crystal structures for 1–9
{[Co3(H2O)4(ptz)2(btec)]·5H2O}n (1).
Complex 1 crystallizes in the triclinic P space group, displays a robust 3D tfz-d framework built from unusual linear trinuclear [Co3(H2O)4(btec)]2+ cations and a pair of ptz− linkers. As shown in Fig. 1a, the trinuclear [Co3(H2O)4(btec)]2+ subunit is centrosymmetric, consisting of three separate CoII atoms with Co1 locating at the crystallographic inversion center, one fully deprotonated btec4− anion with another inversion center, and two pairs of coordinated water molecules. Both crystallographically independent CoII atoms have distorted octahedral geometry (see ESI, Fig. S1),† in which Co1 is equatorially surrounded by four carboxylate O atoms from two different btec4− ligands and axially occupied by two pyrazinyl N atoms from two separate ptz− anions. Instead, Co2 is coordinated by three N atoms from two ptz− ligands, one carboxylate O donor of btec4− anion and two coordinated water molecules, respectively. The bond distances of Co–O are generally shorter than those of Co–N (see Table 2), although they are comparable to those previously reported values.29 Co1, symmetry-related Co2 and Co2A (symmetry code: A = −x, 1 − y, 1 − z) are doubly held together by two carboxylate groups of btec4− in bidentate bridging and monodentate modes to generate a linear trinuclear [Co3(H2O)4(btec)]2+ subunit bearing four terminal water molecules (see Fig. 1a). The Co⋯Co separation within the trinuclear subunit is 5.0432(2) Å, and the bond angle of Co2–Co1–Co2A is 180°.
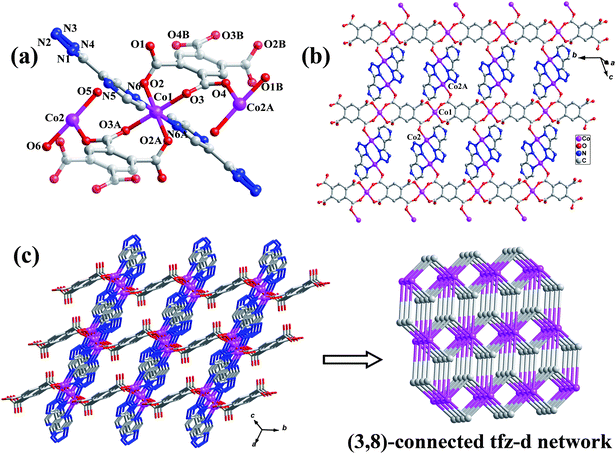 |
| | Fig. 1 (a) The trinuclear CoII subunit of 1 (the centrosymmetric btec4− ligands are shaded differently, and H atoms are omitted for clarity; Symmetry codes: A = −x, 1 − y, 1 − z; B = 1 − x, −y, 1 − z). (b) 2D layer of 1. (c) 3D framework of 1 and its binodal (3,8)-connected tfz-d topological network. | |
Table 2 Selected bond lengths (Å) and angles (°) for 1a
|
Symmetry codes: for 1: #1 −x + 1, −y + 1, −z + 1; #2 −x, −y + 1, −z + 1; #3 −x, −y + 2, −z + 2.
|
| Co(1)–O(2) |
2.0321(16) |
Co(2)–O(5) |
2.071(2) |
| Co(1)–O(3) |
2.0594(17) |
Co(2)–O(4)#2 |
2.0762(17) |
| Co(1)–N(6) |
2.207(2) |
Co(2)–N(2)#3 |
2.110(2) |
| Co(2)–N(5) |
2.207(2) |
Co(2)–N(1) |
2.145(2) |
| Co(2)–O(6) |
2.069(2) |
|
|
| O(2)–Co(1)–O(2)#1 |
180.0 |
O(6)–Co(2)–N(2)#3 |
92.02(9) |
| O(2)–Co(1)–O(3) |
90.84(7) |
O(5)–Co(2)–N(2)#3 |
89.78(9) |
| O(2)#1–Co(1)–O(3) |
89.16(7) |
O(4)#2–Co(2)–N(2)#3 |
91.96(8) |
| O(2)#1–Co(1)–O(3)#1 |
90.83(7) |
O(6)–Co(2)–N(1) |
87.73(8) |
| O(2)–Co(1)–N(6) |
88.33(7) |
O(5)–Co(2)–N(1) |
86.36(8) |
| O(2)#1–Co(1)–N(6) |
91.67(7) |
O(4)#2–Co(2)–N(1) |
171.17(8) |
| O(3)–Co(1)–N(6) |
89.45(7) |
N(2)#3–Co(2)–N(1) |
95.68(8) |
| O(3)#1–Co(1)–N(6) |
90.55(7) |
O(6)–Co(2)–N(5) |
89.81(9) |
| O(3)#1–Co(1)–N(6)#1 |
89.44(7) |
O(5)–Co(2)–N(5) |
87.66(9) |
| O(6)–Co(2)–O(5) |
173.97(9) |
N(2)#3–Co(2)–N(5) |
172.22(8) |
| O(6)–Co(2)–O(4)#2 |
87.59(8) |
N(1)–Co(2)–N(5) |
76.83(8) |
| O(5)–Co(2)–O(4)#2 |
98.11(8) |
|
|
Owing to the presence of centrosymmetric btec4− anions, the trinuclear subunits are covalently extended to 1D ribbon along the crystallographic b-direction. A pair of head-to-tail arranged ptz− ligands present their tetrazinyl N1, N2 and pyrazinyl N5 donors to chelate the Co2 within 1D ribbon and further bridge another Co2 from the parallel ribbons, resulting in a wave-like 2D layer (Fig. 1b). These layers are then covalently stacked together by the coordination bonds between the pyrazinyl N6 of ptz− ligand and Co1 atom to produce a 3D robust MOF (Fig. 1c), which is isostructural to our previously reported [Ni3(H2O)4(ptz)2(btec)]·4H2O}n.24 Thus, the ptz− ligand in 1 presents a tetradentate μ3-κ2N1,N5:κ1N2:κ1N6 chelating-bridging mode to contribute to the covalent 2D and 3D connectivity. Topologically, if the [Co3(H2O)4(COO)4]2+ core and ptz− ligand are considered as two different nodes, 1 can be simplified as a bimodal (3,8)-connected tfz-d net with the (43)2(46·618·84) topology symbol by OLEX.30 On the other hand, the (3,8)-connected framework is microporous with 151.1 Å 3 cavity volume when removing the lattice water molecules.31 The resulting solvent-accessible cavities were estimated to be 18.7% of the unit cell volume.
[Cu2(H2O)4(ptz)2(H2btec)]n (2).
Complex 2 exhibits an infinite 1D chain assembled from the centrosymmetric binuclear [Cu2(H2O)4(ptz)2]2+ nodes and doubly deprotonated H2btec2− connectors. As shown in Fig. 2a, the unique CuII atom has a slightly distorted octahedral structure surrounded by three N donors from two anionic ptz− ligands, one carboxylate O atom from partial deprotonated H2btec2− co-ligand and two coordinated water molecules (see Table 3). Acting as a tridentate chelating-bridging ligand, a pair of ptz− anions present a μ2-κ2N1,N5:κ1N2 binding mode to chelate Cu1 center by tetrazinyl N1 and pyrazinyl N5 donors and simultaneously bridge the centrosymmetric Cu1A by tetrazinyl N2 atom, generating a rigid bimetallic node with the Cu⋯Cu separation of being 4.0599(4) Å. Furthermore, the binuclear [Cu2(H2O)4(ptz)2]2+ nodes are infinitely extended by doubly deprotonated H2btec2− linkers to led to an infinite 1D covalent chain (see Fig. 2a), in which the nearest Cu⋯Cu distance across the H2btec2− co-ligand is 11.0437(10) Å.
 |
| | Fig. 2 (a) View of the 1D chain of 2 (H-atoms were omitted for clarity, symmetry codes: A = −x + 1, −y + 2, −z + 2; B = −x + 2, −y + 2, −z + 1). (b) 2D supramolecular layer of 5 formed by intermolecular O–H⋯O and O–H⋯N hydrogen bonding interactions. | |
Table 3 Selected bond lengths (Å) and angles (°) for 2a
|
Symmetry Codes for 2: #1 −x + 1, −y + 2, −z + 2; #2 −x + 2, −y + 2, −z + 1.
|
| Cu(1)–O(1) |
1.932(2) |
Cu(1)–N(1) |
2.003(2) |
| Cu(1)–N(2)#1 |
2.005(3) |
Cu(1)–N(5) |
2.058(3) |
| Cu(1)–O(5) |
2.316(2) |
Cu(1)–O(6) |
2.645(4) |
| O(1)–Cu(1)–N(1) |
173.06(10) |
O(1)–Cu(1)–N(2)#1 |
91.15(9) |
| N(1)–Cu(1)–N(2)#1 |
94.11(10) |
O(1)–Cu(1)–N(5) |
94.70(9) |
| N(1)–Cu(1)–N(5) |
80.44(10) |
N(2)#1–Cu(1)–N(5) |
172.59(10) |
| O(1)–Cu(1)–O(5) |
86.52(9) |
N(1)–Cu(1)–O(5) |
88.65(10) |
| N(2)#1–Cu(1)–O(5) |
93.33(10) |
N(5)–Cu(1)–O(5) |
91.56(9) |
| N(1)–Cu(1)–O(6) |
85.36(12) |
O(1)–Cu(1)–O(6) |
98.61(11) |
| N(5)–Cu(1)–O(6) |
78.76(17) |
N(2)#1–Cu(1)–O(6) |
95.88(18) |
| O(5)–Cu(1)–O(6) |
169.34(16) |
|
|
In the packing structure of 2, both the undeprotonated carboxylic group and terminal water molecule can act as hydrogen-bond donors to produce the interchain O–H⋯O and O–H⋯N noncovalent interactions with the deprotonated carboxylate group and pyrazinyl N6 acceptors of ptz− ligand (see Table S1), which assemble the separate chains into a 2D supramolecular layer with the nearest interchain Cu⋯Cu separation of 8.2326(11) Å. Additionally, face-to-face π⋯π stacking interactions between interchain pyrazinyl and tetrazinyl rings are further reinforced these 2D layers (Fig. 2b). Furthermore, the adjacent 2D layers are further linked together to form a 3D network through the O5–H5B⋯O2 hydrogen-bonding interactions (see ESI, Fig. S2 and Table S1).†
Of the three ternary metal complexes (1, 2, and previously reported {[Ni3(H2O)4(ptz)2(btec)]·4H2O}n24), the btec4− co-ligand in both 1 and {[Ni3(H2O)4(ptz)2(btec)]·4H2O}n can act as a fully deprotonated hexadentate ligand to aggregate and extend two different trimetallic cores into two isostructural 3D polymers. In contrast, the incomplete deprotonated co-ligand in 2 just behaves as a connector to link dimeric Cu2(ptz)2 subunits into a 1D chain through bidentate bridging coordination mode.
[Hg2(ptz)2Cl2]n (3).
Complex 3 is a trinodal (3,4)-connected 3D framework, showing unusual (6·82)2(6·83·102) topology.30 The asymmetric unit of 3 consists of two crystallographically independent HgII ions, two ptz− ligands, and two Cl− atoms. As shown in Fig. 3a, both HgII atoms are five-coordinated, displaying the slightly distorted trigonal bipyramidal geometry. Hg1 atom is surrounded by one bridging Cl− atom and four N donors (N1, N3C, N5, N12) from three separate ptz− ligands. Instead, the N3Cl2 donor set of Hg2 is completed by three N atoms from two ptz− ligands, one bridging Cl1, and one terminal Cl2 ligand, respectively. The bond lengths of Hg–Cl are generally longer than those of Hg–N (see Table 4), although they are comparable to those previously reported values.32 Two ptz− ligands adopt μ2-κ2N1,N5:κ1N6 and μ3-κ2N1,N5:κ1N3:κ1N6 modes to alternately link Hg1 and Hg2 into a zigzag-chain along the crystallographic c-axis and further extend the adjacent chains into a 2D covalent layer in bc-plane (Fig. 3b). The bridging Cl1 atom then connects the adjacent Hg1 and Hg2 atoms from the adjacent layers into a 3D robust MOF (Fig. 3c). Topologically, when the Hg1, Hg2, and tetradentate ptz− are viewed as three different nodes (three-connected Hg1/Hg2 and four-connected ptz−), the 3D framework of 3 can be simplified into a trinodal (3,4)-connected network with the (6·82)2(6·83·102) topology (Fig. 3c).30
 |
| | Fig. 3 (a) Local coordination environments of HgII atoms in 3 and the coordination mode of the ptz− ligand (H atoms were omitted for clarity; Symmetry codes: A = x, 0.5 − y, 0.5 + z; B = 1 − x, −0.5 + y, 0.5 − z; C = 0.5 −x, −0.5 + y, z; D = x, 0.5 − y, −0.5 + z, E = 0.5 − x, 0.5 + y, z; F = 1 − x, 0.5 + y, 0.5 − z). (b) 2D HgII-ptz layer of 3. (c) 3D framework of 3 and its trinodal (3,4)-connected (6·82)2(6·83·102) topology. | |
Table 4 Selected bond lengths (Å) and angles (°) for 3–5a
|
Symmetry codes for 3: #1 x, −y + 1/2, z + 1/2; #2 −x + 1, y − 1/2, −z + 1/2; #3 −x + 1/2, y − 1/2, z. for 4: #1−x, −y + 2, − z. for 5: #1 −x + 1, −y + 1, −z + 1.
|
|
3
|
| Hg(1)–N(1) |
2.126(6) |
Hg(1)–Cl(1) |
2.3274(1) |
| Hg(1)–N(12) |
2.507(7) |
Hg(1)–N(3)#3 |
2.7743(1) |
| Hg(1)–N(5) |
2.7294(1) |
Hg(2)–Cl(1) |
3.0603(1) |
| Hg(2)–N(7) |
2.111(6) |
Hg(2)–Cl(2) |
2.2901(2) |
| Hg(2)–N(6)#1 |
2.568(7) |
Hg(2)–N(11) |
2.7873(1) |
| N(1)–Hg(1)–Cl(1) |
162.78(17) |
N(1)–Hg(1)–N(12) |
101.4(2) |
| Cl(1)–Hg(1)–N(12) |
95.47(16) |
N(3)#3–Hg(1)–Cl(1) |
106.955(1) |
| N(3)#3–Hg(1)–N(5) |
85.801(1) |
N(3)#3–Hg(1)–N(12) |
87.025(1) |
| N(5)–Hg(1)–Cl(1) |
94.620(1) |
N(5)–Hg(1)–N(12) |
168.981(1) |
| N(5)–Hg(1)–N(1) |
68.911(1) |
N(3)#3–Hg(1)–N(1) |
77.747(1) |
| Cl(1)#2–Hg(2)–N(6)#1 |
78.791(1) |
Cl(1)#2–Hg(2)–N(7) |
84.394(1) |
| N(7)–Hg(2)–Cl(2) |
164.2(2) |
N(7)–Hg(2)–N(6)#1 |
94.3(2) |
| Cl(2)–Hg(2)–N(6)#1 |
101.23(17) |
Cl(2)–Hg(2)–N(11) |
97.599(1) |
| N(11)–Hg(2)–N(7) |
67.990(1) |
N(11)–Hg(2)–N(6) |
142.879(2) |
| Cl(1)#2–Hg(2)–Cl(2) |
100.811(1) |
Cl(1)#2–Hg(2)–N(11) |
128.383(1) |
|
4
|
| Hg(1)–N(7) |
2.057(5) |
Hg(1)–O(1) |
2.657(6) |
| Hg(1)–N(11) |
2.759(7) |
Hg(1)–N(5) |
2.742(1) |
| Hg(1)–N(1) |
2.090(5) |
Hg(2)–N(2) |
2.8428(2) |
| Hg(1)–N(13) |
2.642(5) |
Hg(2)–N(13) |
2.026(5) |
| N(7)–Hg(1)–N(1) |
175.47(19) |
N(13)–Hg(1)–O(1) |
75.41(19) |
| N(7)–Hg(1)–N(13) |
93.79(18) |
N(13)#1–Hg(2)–N(13) |
179.999(2) |
| N(1)–Hg(1)–N(13) |
85.74(17) |
N(2)#1–Hg(2)–N(2) |
180.000 |
| N(7)–Hg(1)–O(1) |
91.1(2) |
N(2)#1–Hg(2)–N(13) |
94.079(3) |
| N(1)–Hg(1)–O(1) |
84.4(2) |
N(13)–Hg(2)–N(2) |
85.921(2) |
|
5
|
| Hg(1)–N(1)#1 |
2.080(4) |
Hg(1)–O(1) |
2.539(4) |
| Hg(1)–N(5) |
2.7082(4) |
|
|
| N(1)#1–Hg(1)–N(1) |
179.999(1) |
N(1)#1–Hg(1)–O(1)#1 |
89.49(17) |
| O(1)#1–Hg(1)–O(1) |
180.000(1) |
N(1)#1–Hg(1)–O(1) |
90.51(17) |
| N(1)–Hg(1)–N(5) |
69.493(3) |
N(1)–Hg(1)–O(1) |
89.49(17) |
| N(5)–Hg(1)–O(1) |
101.250(3) |
N(5)–Hg(1)–O(1)#1 |
78.750(3) |
| N(5)#1–Hg(1)–N(5) |
180 |
N(1)#1–Hg(1)–N(5) |
110.507(4) |
| N(1)–Hg(1)–O(1)#1 |
90.505(3) |
|
|
[Hg3(H2O)2(ptz)4(μ1,1-N3)2]·2H2O (4).
Rather than being polymeric frameworks of 1–3, complex 4 is a discrete trinuclear entity bridged by pairs of N3− and ptz− anions. The fundamental unit of 4 consists of three separate HgII atoms with Hg2 locating at the crystallographic inversion center, two terminal water molecules, a pair of N3− anions, four separate ptz− anions acting as bridging and terminal ligands respectively, and two lattice water molecules. As shown in Fig. 4a, Hg1 is in a distorted octahedron formed by one terminal water molecule and five N atoms from one N3− and two different ptz− ligands. Instead, Hg2 is in a square-planar arrangement to four N atoms from pairs of ptz− and N3− anions (see Table 4).
 |
| | Fig. 4 (a) The discrete trinuclear structure of 4 (H atoms were omitted for clarity; symmetry code: A = −x, −y + 2, −z). (b) 2D supramolecular layer of 4 formed by O–H⋯N hydrogen bonding interactions. | |
Hg2 and Hg1 as well as Hg2 and Hg1A are doubly strengthened by the azide anion and one of the crystallographically independent ptz− ligand containing N1∼N6 to generate linear trinuclear structure with the bond angle of Hg1–Hg2–Hg1A of 180°. The azide anion in trimetallic core adopts an end-on bridging mode and the ptz− ligand acts in a tridentate μ2-κ2N1,N5:κ1N2 fashion. The Hg⋯Hg separation within the trinuclear unit is 3.9459(3) Å. By contrast, the other ptz− anion defined by N7∼N12 only presents its N7 and N11 donors to chelate Hg1 atom in a bidentate μ1-κ2N1,N7 mode. Thus, the two ptz− anions in 4 play different roles (bridging and terminal roles) for the formation of the unusual trinuclear structure. As shown in Fig. 4b, intermolecular O–H⋯N hydrogen-bonding interactions between the coordinated water and ptz− ligands assemble the discrete trinuclear units into a 2D supramolecular layer.
Hg(H2O)2(ptz)2 (5).
Complex 5 without any organic co-ligand exhibits a centrosymmetric mononuclear structure. The unique HgII ion lies on the inversion center and coordinated by four N atoms from a pair of chelating ptz− ligands in an equatorial plane and two water molecules in the axial positions (Fig. 5a), displaying the deformed octahedral coordination geometry (see Table 4). The discrete mononuclear structure is isostructural to various metal complexes with the ptz− ligand, [M(ptz)2(H2O)2] (M = CdII,5 ZnII,2,33 MnII,33,34 FeII,35 CoII,3,36 CuII,3 NiII,37). Each centrosymmetric mononuclear structure is H-bonded with its six neighbours through two pairs of O–H⋯N interactions produced by coordinated water and ptz− ligand, giving rise to a regular 3D hydrogen-bonded network (see Fig. 5b and ESI, Table S1).†
 |
| | Fig. 5 (a) Mononuclear structure of 5 (H atoms were omitted for clarity; symmetry code: A = −x + 1, −y + 1, −z + 1). (b) 3D packing diagram of 5 formed by O–H⋯N hydrogen-bonding interactions. | |
[Cd2(H2O)2(ptz)2(ox)]n (6).
Complex 6 exhibits a 6-connected 3D α-Po-type framework with cationic CdII-oxalate− strands interconnected by bidentate bridging ptz− ligands. The asymmetric unit of 6 consists of one CdII cation, one ptz− anion, half an oxalate dianion with an inversion center, and one coordinated water molecule. As shown in Fig. 6a, the crystallographic independent Cd1 atom is in a distorted octahedron completed by three carboxylate O atoms from two different oxalate dianions and one water molecule in an equatorial plane and two pyrazinyl N atoms from two symmetry-related ptz− anions locating at the axial positions (see Table 5). Each centrosymmetric oxalate co-ligands adopts a μ4-κ2O1′,O2:κ1O2;κ2O1,O2′:κ1O2′ chelating-bridging mode to coordinate with four crystallographically equivalent CdII atoms. As a result, an infinite CdII-oxalate strand is generated along the crystallographic a-axis with the nearest Cd⋯Cd distance of 3.8709(3) Å (see Fig. 6b). Furthermore, the ptz− ligand serves as a bridging μ2-κ1N1:κ1N6 connector to link the antiparallel CdII-oxalate strands into a 3D architecture (see Fig. 6c). This is the first time that the μ2-κ1N1:κ1N6 coordination mode of the ligand is observed in ptz−−based metal complexes. Topologically, if the bimetallic [Cd2(H2O)2]4+ cation bridged by carboxylate O2 of oxalate co-ligand is considered as a node and the bridged oxalate and ptz− ligands as the linkers, the 3D framework of 6 can be simplified into a 6-connected α-Po-type structure with the (412·63) topology symbol (see Fig. 6c).30
 |
| | Fig. 6 (a) Local environment of CdII atom in 6 (H-atoms were omitted for clarity, symmetry codes: A = x − 1, y, z; B = −x + 0.5, y + 0.5, −z + 1.5; C = 2 − x, 1 − y, 2 − z). (b) The linkage of the oxalate-CdII ribbon. (c) 3D framework of 6 and its 6-connected α-Po-type topological structure. | |
Table 5 Selected bond lengths (Å) and angles (°) for 6–9a
|
Symmetry codes for 6: #1 x − 1, y, z; #2 −x + 1/2, y + 1/2, − z + 3/2; #3 −x + 2, −y + 1, −z + 2. For 7: #1 −x, −y, −z, #2 − x, y−1/2, − z + 1/2. For 8: #1 − x + 1, − y + 1, −z, #2 −x, −y + 1, −z. For 9: #1 −x + 1, −y, −z + 1; #2 −x + 2, −y, −z + 1.
|
|
6
|
| Cd(1)–N(1) |
2.283(2) |
Cd(1)–O(2)#1 |
2.3121(16) |
| Cd(1)–O(3) |
2.3150(18) |
Cd(1)–O(1) |
2.3682(16) |
| Cd(1)–N(6)#2 |
2.369(2) |
Cd(1)–O(2)#3 |
2.4007(16) |
| N(1)–Cd(1)–O(2)#1 |
96.89(7) |
N(1)–Cd(1)–O(3) |
100.68(7) |
| O(2)#1–Cd(1)–O(3) |
143.14(6) |
N(1)–Cd(1)–O(1) |
83.21(6) |
| O(2)#1–Cd(1)–O(1) |
137.85(6) |
O(3)–Cd(1)–O(1) |
76.68(6) |
| N(1)–Cd(1)–N(6)#2 |
177.86(7) |
O(2)#1–Cd(1)–N(6)#2 |
81.18(6) |
| O(3)–Cd(1)–N(6)#2 |
81.44(7) |
O(1)–Cd(1)–N(6)#2 |
97.59(7) |
| N(1)–Cd(1)–O(2)#3 |
86.42(6) |
O(2)#1–Cd(1)–O(2)#3 |
69.57(6) |
| O(3)–Cd(1)–O(2)#3 |
143.26(6) |
O(1)–Cd(1)–O(2)#3 |
68.37(5) |
| N(6)#2–Cd(1)–O(2)#3 |
92.03(6) |
|
|
|
7
|
| Cd(1)–N(2)#1 |
2.298(3) |
Cd(1)–O(1) |
2.359(3) |
| Cd(1)–N(6)#2 |
2.412(3) |
Cd(1)–N(1) |
2.423(2) |
| Cd(1)–N(5) |
2.427(3) |
Cd(1)–I(1) |
2.7871(8) |
| N(2)#1–Cd(1)–O(1) |
92.16(10) |
N(2)#1–Cd(1)–N(6)#2 |
93.55(10) |
| O(1)–Cd(1)–N(6)#2 |
162.42(9) |
N(2)#1–Cd(1)–N(1) |
88.53(9) |
| O(1)–Cd(1)–N(1) |
79.43(9) |
N(6)#2–Cd(1)–N(1) |
84.11(9) |
| N(2)#1–Cd(1)–N(5) |
158.45(9) |
O(1)–Cd(1)–N(5) |
84.73(9) |
| N(6)#2–Cd(1)–N(5) |
83.86(10) |
N(1)–Cd(1)–N(5) |
69.93(9) |
| N(2)#1–Cd(1)–I(1) |
105.31(7) |
O(1)–Cd(1)–I(1) |
96.55(6) |
| N(6)#2–Cd(1)–I(1) |
97.94(6) |
N(1)–Cd(1)–I(1) |
165.79(6) |
| N(5)–Cd(1)–I(1) |
96.23(6) |
|
|
|
8
|
| Cd(1)–Br(1)#1 |
2.6992(11) |
Br(1)–Cd(1) |
2.7990(12) |
| Cd(1)–N(2)#2 |
2.300(6) |
Cd(1)–N(1) |
2.356(5) |
| Cd(1)–O(1) |
2.145(2) |
Cd(1)–N(5) |
2.396(6) |
| N(2)#2–Cd(1)–N(1) |
93.11(19) |
N(2)#2–Cd(1)–O(1) |
89.6(2) |
| N(1)–Cd(1)–O(1) |
87.44(19) |
N(2)#2–Cd(1)–N(5) |
162.4(2) |
| N(1)–Cd(1)–N(5) |
70.99(19) |
O(1)–Cd(1)–N(5) |
82.29(19) |
| N(2)#2–Cd(1)–Br(1)#1 |
98.72(14) |
N(1)–Cd(1)–Br(1)#1 |
167.68(14) |
| O(1)–Cd(1)–Br(1)#1 |
89.21(13) |
N(5)–Cd(1)–Br(1)#1 |
96.82(14) |
| N(2)#2–Cd(1)–Br(1) |
90.19(15) |
N(1)–Cd(1)–Br(1) |
90.80(14) |
| O(1)–Cd(1)–Br(1) |
178.21(13) |
N(5)–Cd(1)–Br(1) |
97.42(14) |
| Br(1)#1–Cd(1)–Br(1) |
92.58(3) |
|
|
|
9
|
| Cd(1)–N(2)#1 |
2.302(4) |
Cd(1)–N(5) |
2.392(4) |
| Cd(1)–N(1) |
2.361(4) |
Cd(1)–Cl(1) |
2.5812(18) |
| Cd(1)–O(1) |
2.371(4) |
Cd(1)–Cl(1)#2 |
2.672(2) |
| N(2)#1–Cd(1)–N(1) |
92.98(14) |
N(2)#1–Cd(1)–O(1) |
88.86(14) |
| N(1)–Cd(1)–O(1) |
88.01(14) |
N(2)#1–Cd(1)–N(5) |
161.85(14) |
| N(1)–Cd(1)–N(5) |
71.10(14) |
O(1)–Cd(1)–N(5) |
82.10(13) |
| N(2)#1–Cd(1)–Cl(1) |
98.48(11) |
N(1)–Cd(1)–Cl(1) |
168.29(11) |
| O(1)–Cd(1)–Cl(1) |
89.85(11) |
N(5)–Cd(1)–Cl(1) |
97.21(11) |
| N(2)#1–Cd(1)–Cl(1)#2 |
91.42(12) |
N(1)–Cd(1)–Cl(1)#2 |
91.64(11) |
| O(1)–Cd(1)–Cl(1)#2 |
179.57(11) |
N(5)–Cd(1)–Cl(1)#2 |
97.54(11) |
| Cl(1)–Cd(1)–Cl(1)#2 |
90.44(6) |
|
|
[Cd(H2O)(ptz)I]n (7).
Complex 7 exhibits a 2D layered structure with the centrosymmetric binuclear [Cd2(H2O)2I2]2+ cations extended by pairs of anionic ptz− linkers. As shown in Fig. 7a, the fundamental subunit is a centrosymmetric dimer constructed from pairs of CdII atoms, ptz− anions, coordinated water molecules and I− ligand. The CdII atom is in a slightly distorted octahedral geometry formed by four N atoms from three separate ptz− ligands, one terminal I− anion and one coordinated water molecule. Each ptz− acts as a tetradentate μ3-κ2N1,N5:κ1N2:κ1N6 chelating-bridging ligand to chelate one centrosymmetric Cd1 center and simultaneously bridge two other symmetry–related Cd1 atoms. The adjacent 2D layers are further assembled into a 3D network through the O1–H1B⋯I1 and O1–H1A⋯N4 hydrogen-bonding interactions between aqua and I−/ptz− ligand (see ESI, Fig. S3).†
 |
| | Fig. 7 (a) The centrosymmetric binuclear subunit and the local coordination environment of the CdII atom of 7 (H-atoms were omitted for clarity, symmetry codes: A = −x, −y, −z; B = −x, y − 0.5, −z + 0.5). (b) 2D covalent layer of 7. | |
{[Cd(H2O)(ptz)Br]·H2O}n (8) and {[Cd(H2O)(ptz)Cl]·H2O}n (9).
By changing the co-ligand from I− with larger atomic radius to comparatively small Br− or Cl− atom, the 2D layered structure of 7 is decreased to 1D chains with the binuclear [Cd2(H2O)2(ptz)2]+ cations alternately propagated by bridged Br− anion for 8 or Cl− anion for 9, respectively. The substructure of the bimetallic [Cd2(H2O)2(ptz)2]+ cation is also centrosymmetric dimer with the CdII atom in a slightly distorted octahedral geometry surrounded by three N donors from a pair of centrosymmetric ptz− ligands, two bridging X atoms and one water molecule (see Fig. 8a, Tables 5). Different from the chelating-bridging role of the ptz− ligand in 7, the core ligand in 8 and 9 only chelates two symmetry-related Cd1 and Cd1A to generate the binuclear subunit, rather than extending the high dimensional framework of the target complexes. Instead, pairs of halide anions doubly bridge the neighbouring binuclear subunits to lead to an infinite chain (see Fig. 8a). The Cd⋯Cd distance across the bridging halide anion is 3.8001(4) for 8 and 3.7011(2) Å for 9, which is 0.83 and 0.72 Å shorter than that chelated by ptz− ligand. As shown in Fig. 8b, the chain-based structure is expanded into 3D supramolecular network through strong O–H⋯N hydrogen-bonding interactions between coordinated water and ptz− ligand (see ESI, Table S1).†
 |
| | Fig. 8 (a) 1D chain of 8 or 9 formed by bimetallic nodes and bridging halide linkers (H-atoms were omitted for clarity, symmetry codes: A = 1 − x, −y, 1 − z; B = 2 − x, −y, 1 − z). (b) 3D supramolecular network of 8 or 9 assembled from interchain O–H⋯N hydrogen bonding interactions. | |
Notably, although 8 and 9 possess the analogous covalent chain-based skeleton, the intermolecular weak interactions between lattice water and organic framework are different from each other. Lattice water can produce three-fold hydrogen-bonding interactions with the framework of 8 (two O–H⋯N and one O–H⋯O interactions, see ESI, Table S1).† In contrast, only one O–H⋯O hydrogen-bonding interaction can be observed in 9 (see ESI, Fig. S4 and Table S1).† On the other hand, comparisons of 7 bearing terminal I− co-ligand with 8 and 9 bearing bridging Br− or Cl− auxiliary ligand reveal that the presence of the competing binding behaviour between the mixed-ligands due to the increase of halide radius.
Binding preference of the 5-(pyrazinyl)tetrazolate ligand in metal complexes
The 5-(pyrazinyl)tetrazolate ligand can potentially afford six different metal binding sites or their variable combinations, since it is structurally related to a tetrazolate ring connected with a pyrazinyl ring. Indeed, it has become a versatile ligand for bridging or chelating different metal ions. Up to date, eight different binding modes by the ligand (I: μ1-κ2N1,N5, II: μ2-κ1N1:κ1N6, III: μ2-κ1N2:κ1N4, IV: μ2-κ2N1,N5:κ1N6, V: μ2-κ2N1,N5:κ1N2, VI: μ3-κ2N1,N5:κ1N2:κ1N6, VII: μ3-κ2N1,N5:κ1N3:κ1N6, VIII: μ4-κ1N1:κ1N2:κ1N3:κ1N6)2–4,15,34,38 have been observed, which were summarized in Fig. 9. The ptz− anion is repeatedly observed as a bidentate, tridentate and even tetradentate ligand to chelate and/or bridge different metal ions. Undoubtedly, such different coordination modes are significantly influenced by the presence of the different co-ligands and the metal centers. When a co-ligand was introduced in the ternary systems, cooperative binding can occur or bonding competition can be observed between the bridging, chelating and/or bridging-chelating ptz− core-ligand and the terminal or bridging co-ligands. For example, ptz− anion in complexes 2, 4, 5, 8, and 9 can only act as chelating/chelating-bridging ligand to complete the mono-, bi-, and tri-metallic subunit, respectively. By contrast, in 6 it just behaves as a bridging ligand to connect the adjacent CdII-oxalate ribbon. Interestingly, in complexes 1, 3, and 7, the ptz− anion also serves as chelating-bridging ligand to extend the dimensionalities of the covalent frameworks. Additionally, the remained vacant N sites from the polydentate ptz− ligand can serve as good hydrogen-bonding acceptors, which can significantly produce multiple noncovalent interactions to contribute to the architectural dimensionality. Such supramolecular interactions have been previously discussed in more detail.2–3,15
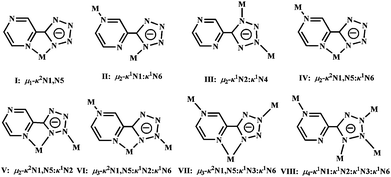 |
| | Fig. 9 Summary of the binding modes for anionic ptz− ligand in the metal complexes. | |
For the three HgII-based polymers, complex 3 without any lattice molecules is thermally stable up to 150 °C. Then an obvious weight-loss process is observed until 305 °C, corresponding to the decomposition of ptz− ligand, vaporization of HgCl2 and the formation of mercury (expt. 74.3% calcd 73.8%). Upon further heating, the residual mercury was slowly evaporated.38 The TG curves of 4 and 5 are similar with each other. The first stage between 100 °C and 200 °C for 4 (expt. 5.7% calcd 5.4%) as well as between 77 °C and 128 °C for 5 (obs. 6.7%, calcd 6.8%) is due to the complete loss of water molecules. The second obvious weight-loss from 200 °C to 277 °C for 4 and from 237 °C to 267 °C for 5 is ascribed to the removal of organic ligands and mercury, respectively. Upon further heating to 800 °C, the residual mercury gradually evaporated.
The thermal decomposition profiles of 6–9 display consistently two-stage weight-loss processes for free and bound water molecules from 99 to 302 °C and the mixed-ligands from 317 to 633 °C, respectively. Surprisingly, the initial temperature (250 °C) of the first stage of 6 (250–302 °C; expt. 5.5%, calcd 5.6%) is much more stable by 140 °C than those of 7 (120 °C corresponding to the first stage between 120 and 154 °C, expt. 4.8%, calcd 4.5%), 8 (99 °C corresponding to the first stage between 99 and 180 °C, expt. 9.8%, calcd 9.6%) and 9 (106 °C corresponding to the first stage between 106 and 180 °C, expt. 9.2%, calcd 10.9%). The final product is CdO for 6 (expt. 40.0%, calcd 39.9%) and 9 (expt. 18.4%, calcd 19.4%), and Cd(CN)2 for 7 (expt. 20.4%, calcd 20.3%) and 8 (expt. 22.6%, calcd 21.9%).
Luminescence compounds are of great current interest because of their various applications in chemical sensors, photochemistry and structure electroluminescence (EL) displays and light-emitting diodes (LEDs).39 Thus, the solid state emission spectra of all the resulting complexes except 2 were measured to explore their potential optical properties. As shown in Fig. 10, upon excitation at 375 nm for 1 and 3–4 or at 385 nm for four CdII-ptz-based complexes, only one emission at 422 nm for both 1 and 3, 425 nm for 4 and 427 nm for both 5 and 9, 428 nm for 6 and 432 nm for 8, was observed, which display slightly variable intensity. By contrast, complex 7 presents two moderate emissions at 423 and 434 nm, respectively. Considering that the isolated neutral ptz ligand can display a strong blue fluorescent emission band at 457 nm upon excitation at 357 nm,15 the emission located ca. 425 nm for all the complexes examined herein should be assigned to the intraligand transition of ptz− ligand upon cation binding. The slight shift of the emission is probably due to the differences of anions and coordination environment around the central metal ions.2,40 The low energy emission at 434 nm for 7 might come from interligand transfer between ptz− and I− anions.
Magnetic behaviour of 2
As shown in Fig. 11, the χMT value for per CuII ion of 2 is 0.452 cm3 K mol−1 at room temperature, which is slightly greater than that (0.42 cm3 K mol−1) for a single CuII ion with S = 1/2 and g = 2.1. The value of χMT decreases to a minimum value 0.044 cm3 K mol−1 as the temperature decreases to 7 K. Then it increases up to 0.060 cm3 K mol−1 at 4 K. On the other hand, the χMvs. T curve provides more information about the magnetic behaviour of 2: the χM value of 0.0015 cm3 mol−1 at room temperature increases as the temperature decreases, arriving at a maximum value of 0.0105 cm3 mol−1 at 22 K; χM then decreases practically to 0.0056 cm3 mol−1 at 8 K before increasing a value of 0.0150 cm3 mol−1 at 4 K. This behaviour clearly indicates a typical antiferromagnetic coupling and the presence of a small amount of paramagnetic impurities.41 Considering the magnetic exchange interaction occurs between the CuII ions within the ptz−−bridged dimer and between the two adjacent dimers across the bridged H2btec2− co-ligand, the best fits were obtained by using the eqn (1) for S = 1/2, where ρ is defined as the molar fraction of the paramagnetic impurity in 2, J is the coupling constant within the dimer, zJ′ is the coupling constant between two dimers, and the other symbols have the usual meanings.
| |  | (1) |
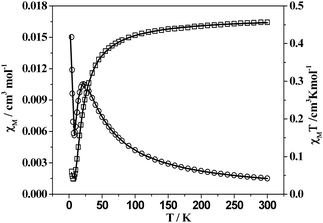 |
| | Fig. 11 Plots of χM (○) and χMT (□) vs.T for 2. Solid lines correspond to the best fit indicated in the text. | |
The results of the best fit were g = 2.19, J = −13.13 cm−1, zJ′ = −0.00018 cm−1, ρ = 0.0335, with r = 2.6 × 10−4, where r is the agreement factor defined as Σ[(χMT)obsd − (χMT)calcd]2/Σ[(χMT)2obsd. Obviously, the difference between J and zJ′ suggests the magnetic superexchange pathway by double ptz− bridges is stronger than that by H2btec2− linker due to the relatively shorter intermetallic distance (4.0599(4) vs. 11.0437(10) Å). Additionally, the coupling constant of 2 is considerable smaller than those of binuclear CuII complexes linked by μ1,2-1,2,4-triazole (−118 < J < −97), which are structurally related with the coordination behaviour of the CuII ions.42 In 2 the unpaired electron of the CuII occupies a magnetic orbital of d(x2 − y2) symmetry, which is pointing towards the equatorially coordinating tetrazolate N atoms. Thus, the overlap between the magnetic orbital of the CuII ion and the σ-orbitals of the tetrazolate N atom significantly determine the strength of the magnetic coupling. The more these orbitals are directed towards each other the larger the overlap and therefore the antiferromagnetic interaction. Since the asymmetric bridging mode of ptz− ligand in 2 led to the bond angles of Cu1–N1–N2 and Cu1A–N2–N1 of 140.7(2) and 124.38(19)°, which allowed a poor overlap than those with the symmetrical CuII–N–N–CuII structures42 and reasonably contributed a weak coupling constant of 2.
Conclusions
In summary, by selectivly introducing functional auxiliary ligands with different size and binding modes and metal ions with different electron configuration, nine mixed-ligand metal complexes with in situ formed ptz− core-ligand were presented, in which HgII ion became the first example that can be used as Lewis acid catalyst in situ reactions. Compared with the reported binary ptz−−based complexes, the favourably synergistic/competing binding ability contributed from the chelating-bridging ptz− core-ligand and bridging/terminal co-ligands has essentially induced intriguing mono-, bi-, and tri-metallic-core-based coordination architecture ranged from separate 0D entities to infinite 3D frameworks. Furthermore, these target complexes exhibit weak antiferromagnetic coupling behaviour for CuII-based polymer as well as ptz−−based strong photoluminescence for the other complexes.
Acknowledgements
This present work was financially supported by the National Natural Science Foundation of China (20703030, 20871092 and 20973125), the Key Project of Chinese Ministry of Education (grant no. 209003), the Program for New Century Excellent Talents in University (NCET-08-0914), and the National Science Foundation of Tianjin (Grants 10JCZDJC21600 and 10JCYBJC04800) which are gratefully acknowledged.
References
-
(a) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS;
(b) S. G. Telfer and R. Kuroda, Coord. Chem. Rev., 2003, 242, 33 CrossRef CAS;
(c) S. A. Barnett and N. R. Champness, Coord. Chem. Rev., 2003, 246, 145 CrossRef CAS;
(d) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS.
- Z. Li, M. Li, X.-P. Zhou, T. Wu, D. Li and S. W. Ng, Cryst. Growth Des., 2007, 7, 1992 CrossRef CAS.
- M. A. M. Abu-Youssef, F. A. Mautner, A. A. Massoud and L. Öhrström, Polyhedron, 2007, 26, 1531 CrossRef CAS.
- J. Luo, X.-R. Zhang, L.-L. Cui, W.-Q. Dai and B. S. Liu, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m614 CrossRef.
- W.-D. Song and D.-L. Xi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m2841 CrossRef.
- A. Rodríguez-Diéguez and E. Colacio, Chem. Commun., 2006, 4140 RSC.
- J.-Y. Zhang, Q. Yue, Q.-X. Jia, A.-L. Cheng and E.-Q. Gao, CrystEngComm, 2008, 10, 1443 RSC.
-
(a) R.-G. Xiong, X. Xue, H. Zhao, X.-Z. You, B. F. Abrahams and Z.-L. Xue, Angew. Chem., Int. Ed., 2002, 41, 3800 CrossRef CAS;
(b) H. Zhao, Z.-R. Qu, H.-Y. Ye and R.-G. Xiong, Chem. Soc. Rev., 2008, 37, 84 RSC.
- P. Lin, W. Clegg, R. W. Harrington and R. A. Henderson, Dalton Trans., 2005, 2388 RSC.
- W. Ouellette, H.-X. Liu, C. J. O'Connor and J. Zubieta, Inorg. Chem., 2009, 48, 4655 CrossRef CAS.
- W.-C. Song, J.-R. Li, P.-C. Song, Y. Tao, Q. Yu, X.-L. Tong and X.-H. Bu, Inorg. Chem., 2009, 48, 3792 CrossRef CAS.
- Y. Chen, Z.-G. Ren, H.-X. Li, X.-Y. Tang, W.-H. Zhang, Y. Zhang and J.-P. Lang, J. Mol. Struct., 2008, 875, 339 CrossRef CAS.
- Z.-R. Qu, H. Zhao, X.-S. Wang, Y.-H. Li, Y.-M. Song, Y.-J. Liu, Q. Ye, R.-G. Xiong, B. F. Abrahams, Z.-L. Xue and X.-Z. You, Inorg. Chem., 2003, 42, 7710 CrossRef CAS.
- D.-S. Liu, G.-S. Huang, C.-C. Huang, X.-H. Huang, J.-Z. Chen and X.-Z. You, Cryst. Growth Des., 2009, 9, 5117 CrossRef CAS.
- Y. Tao, J.-R. Li, Q. Yu, W.-C. Song, X.-L. Tong and X.-H. Bu, CrystEngComm, 2008, 10, 699 RSC.
- E.-C. Yang, H. K. Zhao, B. Ding, X. G. Wang and X. J. Zhao, Cryst. Growth Des., 2007, 7, 2009 CrossRef CAS.
- E.-C. Yang, H. K. Zhao, Y. Feng and X. J. Zhao, Inorg. Chem., 2009, 48, 3511 CrossRef CAS.
- E.-C. Yang, Y.-N. Chan, H. Liu, Z.-C. Wang and X.-J. Zhao, Cryst. Growth Des., 2009, 9, 4933 CrossRef CAS.
- E.-C. Yang, Q.-Q. Liang and P. Wang, Inorg. Chem. Commun., 2009, 12, 211 CrossRef CAS.
- E.-C. Yang, Z.-Y. Liu, X.-G. Wang, S. R. Batten and X.-J. Zhao, CrystEngComm, 2008, 10, 699 RSC.
- E.-C. Yang, Q.-Q. Liang, X.-G. Wang and X.-J. Zhao, Aust. J. Chem., 2008, 61, 813 CrossRef CAS.
-
SAINT; Bruker AXS: Madison, WI, 1998 Search PubMed.
-
(a)
G. M. Sheldrick. SHELXL–97Program for X-ray Crystal Structure Refinement; Göttingen University: Göttingen, Germany, 1997 Search PubMed;
(b)
G. M. Sheldrick. SHELXS–97Program for X-ray Crystal Structure Solution; Göttingen University: Göttingen, Germany, 1997 Search PubMed.
- Y. Feng, E.-C. Yang, M. Fu and X.-J. Zhao, Z. Anorg. Allg. Chem., 2010, 636, 253 CrossRef CAS.
-
K. Nakamoto, Infrared and Raman Spectra of Inorganicand Coordination Compounds, fourth ed., Wiley Press, 1986 Search PubMed.
- X.-Y. Wang, L. Wang, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2006, 128, 674 CrossRef CAS.
- T.-F. Liu, D. Fu, S. Gao, Y.-Z. Zhang, H.-L. Sun, G. Su and Y.-J. Liu, J. Am. Chem. Soc., 2003, 125, 13976 CrossRef CAS.
- Y.-Z. Zhang, H.-Y. Wei, F. Pan, Z.-M. Wang, Z.-D. Chen and S. Gao, Angew. Chem., Int. Ed., 2005, 44, 5841 CrossRef CAS.
-
(a) W. D. Junior, H. J. N. Ishley and R. R. Whittle, Inorg. Chem., 1982, 21, 3270 CrossRef;
(b) J. Costamagna, F. Caruso, M. Rossi, M. Campos, J. Canales and J. Ramirez, J. Coord. Chem., 2001, 54, 247 CrossRef CAS.
- O. V. Dolomanov, A. J. Blake, N. R. Champness and M. J. Schröder, J. Appl. Crystallogr., 2003, 36, 1283 CrossRef CAS.
-
A. L. Spek, PLATONa multipurpose crystallographic tool, Utrecht University, Utrecht, The Netherlands, 2001 Search PubMed.
-
(a) A. Morsali and L. G. Zhu, Helv. Chim. Acta, 2006, 89, 81 CrossRef CAS;
(b) A. G. Orpen, L. Brammer, F. H. Allen, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, S1 RSC.
- J. Luo, X.-R. Zhang, L.-L. Cui, W.-Q. Dai and B.-S. Liu, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m614 CrossRef.
- S.-W. Peng, Y.-L. Miao and W.-D. Song, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 63, m253 CrossRef.
- H. Deng, Y.-C. Qiu, R.-H. Zeng and F. Sun, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m450 CrossRef.
- R.-H. Zeng, Y.-C. Qiu, Z.-H. Liu, Y.-H. Li and H. Deng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m1591 CrossRef.
- S.-D. Fan and J.-T. Liu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2034 CrossRef.
-
(a) S. Stagni, E. Orselli, A. Palazzi, L. D. Cola, S. Zacchini, C. Femoni, M. Marcaccio, F. Paolucci and S. Zanarini, Inorg. Chem., 2007, 46, 9126 CrossRef CAS;
(b) J.-T. Liu, S.-D. Fan and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m1652 CrossRef.
-
(a) Q. Wu, M. Esteghamatian, N. X. Hu, Z. D. Popovic, G. Enright, Y. Tao, M. D'Iorio and S. Wang, Chem. Mater., 2000, 12, 79 CrossRef CAS;
(b) J. E. McGarrah, Y. J. Kim, M. Hissler and R. Eisenberg, Inorg. Chem., 2001, 40, 4510 CrossRef CAS;
(c) G. De Santis, L. Fabbrizzi, M. Licchelli, A. Poggi and A. Taglietti, Angew. Chem., Int. Ed. Engl., 1996, 35, 202 CrossRef.
-
(a) V. W. W. Yam and K. K. W. Lo, Chem. Soc. Rev., 1999, 28, 323 RSC;
(b) B.-D. Lourdes, N. R. David and C.-C. Mercedes, Chem. Soc. Rev., 2007, 36, 993 RSC.
- A. Escuer, M. Font-Bardía, S. S. Massoud, F. A. Mautner, E. Peñalba, X. Solans and R. Vicente, New J. Chem., 2004, 28, 681 RSC.
-
(a) A. Bencini, D. Gatteschi, C. Zanchini, J. G. Haasnoot, R. Prins and J. Reedijk, J. Am. Chem. Soc., 1987, 109, 2926 CrossRef CAS;
(b) R. Prins, P. J. M. W. L. Birker, J. G. Haasnoot, G. C. Verschoor and J. Reedijk, Inorg. Chem., 1985, 24, 4128 CrossRef CAS;
(c) W. M. E. K. Oudenniel, R. A. G. De Graaff, J. G. Haasnoot, R. Prins and J. Reedijk, Inorg. Chem., 1989, 28, 1128 CrossRef CAS;
(d) P. M. Slangen, P. J. van Koningsbruggen, J. G. Haasnoot, J. Jansen, S. Gorter, J. Reedijk, H. Kooijman, W. J. J. Smeets and A. L. Spek, Inorg. Chim. Acta, 1993, 212, 289 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray data in CIF format, TG curves for 1–9 and additional figures and tables. CCDC reference numbers 756331–756339 for 1–9. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c000586j |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here. 
