 Open Access Article
Open Access ArticleTaming functionality: easy-to-handle chiral phosphiranes†
Arne
Ficks
,
Ivan
Martinez-Botella
,
Beverly
Stewart
,
Ross W.
Harrington
,
William
Clegg
and
Lee J.
Higham
*
School of Chemistry, Newcastle University, Bedson Building, Newcastle Upon Tyne, NE1 7RU, UK. E-mail: lee.higham@ncl.ac.uk; Fax: (+44) 191 222 6929; Tel: (+44) 191 222 5542
First published on 24th June 2011
Abstract
Enantiopure chiral phosphiranes possessing a binaphthyl backbone demonstrate remarkable thermal stability, are highly resistant to air-oxidation and are effective ligands in catalytic asymmetric hydrosilylations.
The phosphiranes are heterocycles which possess a three-membered phosphorus-containing ring which is highly strained as a result of the small sum of bond angles at the phosphorus atom (∑(P) <260°).1 The resultant greater s-character in the pyramidalised structure is considered to manifest itself in the form of lowered HOMO and LUMO energy levels, conferring upon the ligand a lower σ-donor, but better π-acceptor electronic profile. Such characteristics are desirable because they offer the opportunity to synthesise ligand libraries for catalysis with a different window of activity from that offered by the more typical PR3 reagents. These intriguing molecules are still overlooked because of their tendency to be unstable:1b,2Phenylphosphirane I (Fig. 1) is stable at 0 °C for a month but undergoes decomposition at higher temperatures.3,4 Bulky substituents around the phosphorus lend stability to the ring; thus II is a distillable liquid5 and III is a colourless crystalline solid.6 The other strategy one can employ to stabilise a phosphirane (apart from metal coordination) is to incorporate the three-membered ring into an elaborate polycyclic structure. This niche approach was used to prepare the elegant BABAR-Phos IV series of phosphiranes which resist decomposition.7 Phosphirano-[1,2]-thiaphospholes V have [a]-annelated bicyclic structures, isomerising in their tungsten complexes over the temperature range 25–80 °C.8
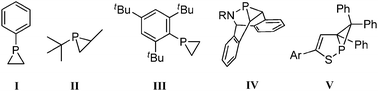 | ||
| Fig. 1 Phosphiranes of varying thermal stability. I is unstable above RT; steric protection (II, III) and ring fusion aid stability (IV, V). | ||
We recently reported an electronic stabilising effect whereby increasing the amount of π-conjugation on the backbone of primary phosphines allowed us to prepare the novel air-stable, chiral derivatives (S)-1a and (R)-1b.9 We were keen therefore to use these precursors to prepare chiral phosphiranes, of which there are very few reports,10 and to examine their properties, structure and reactivity. Following the synthetic method of Kubiak and co-workers,11 (S)-1a and (R)-1b were treated with two equivalents of methyllithium followed by dichloroethane, and thus we were able to prepare the asymmetric compounds (S)-2a and (R)-2b in high yields (Scheme 1).
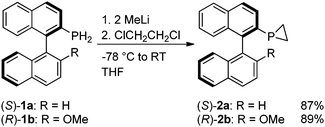 | ||
| Scheme 1 Synthetic procedure for (S)-2a and (R)-2b. | ||
The 31P NMR spectroscopic data for the two phosphiranes are consistent with those found for other members of this class, showing a characteristic resonance at very high field (δ = −235.0 ppm for (S)-2a, −235.4 for (R)-2b). The 1H NMR spectrum shows four distinctive peaks for the protons on the heterocycle, with a complicated fine-structure arising from coupling to each other, coupling to the nearest aromatic proton, and coupling to the phosphorus nucleus. The two proton signals at lower field (endoprotons) both show NOE correlations to the aromatic proton in the 3-position. Thus the phosphirane unit is rotating in solution, an observation further rationalised by quantum chemical calculations.†COSY NMR experiments revealed that the 2J-coupling of the phosphorus nucleus to the two exoprotons is significantly larger (18.0/18.1 Hz in (S)-2a/(R)-2b) than the coupling to the endoprotons (1.8/2.0 Hz in (S)-2a/(R)-2b). The latter smaller coupling was thereby not fully resolvable in the 31P NMR spectra. The 13C NMR spectrum also demonstrates the diastereotopic nature of the phosphirane-ring carbon atoms; two individual resonances are observed although the phosphorus–carbon coupling constants within the phosphirane ring are equivalent (1JPC = 40.3 Hz for (S)-2a; 1JPC = 40.1 Hz for (R)-2b).
Two important features of our novel phosphiranes lie in their high thermal stability and their excellent resistance to air-oxidation. Remarkably (S)-2a and (R)-2b showed no sign of decomposition when heated overnight in refluxing toluene (under nitrogen). Nor was there any evidence of decomposition when the compounds were left open to the atmosphere in chloroform solution for seven days. In fact, in the solid state they can be stored in air over several months without problems. Treatment of the phosphiranes by peroxides in reagent-grade tetrahydrofuran does oxidise the compounds, but this process takes place via a different mechanistic process than the oxidation by air.12 A full rationale for this behaviour will be provided elsewhere, as part of our larger study on the air-stability or otherwise of the phosphine class of compounds in general.13
This remarkable behaviour led us to further investigate the reactivity of the phosphiranes. It has previously been reported that 2,6-dimethoxyphenylphosphirane reacts readily with sulfur at room temperature;14 (S)-2a and (R)-2b gave no reaction under these conditions, nor were any differences observed when the samples were heated at 50 °C in toluene for two hours. However, both (S)-2a and (R)-2b reacted with the strong methylating agent methyl trifluoromethanesulfonate which led to (S)-3a and (R)-3b in a quantitative conversion (Scheme 2). The same reactivity pattern was reported for 1-phenylphosphirane,15 while the stabilised iPrBABARphos phosphirane ligand was found to be unreactive in this conversion,7a further highlighting reactivity differences within the phosphirane class. The 31P NMR spectra showed single peaks at −101.2 ppm for (S)-3a and −102.0 ppm for (R)-3b, whilst in the 1H NMR spectrum, the newly introduced methyl group appears as a doublet for both compounds at 1.60 ppm for (S)-3a (2JPH = 17.9 Hz) and at 1.63 ppm for (R)-3b (2JPH = 18.3 Hz).
 | ||
| Scheme 2 Synthetic procedure for (S)-3a and (R)-3b. | ||
In order to gain further insight into the geometry and electronic nature of the phosphirane group, we undertook a number of quantum chemical calculations of (S)-2a and (R)-2b at the B3LYP/6-31G* level of theory. The HOMO and LUMO energy levels of (S)-2a and (R)-2b were calculated and compared to their unstrained MOP derivatives, 2-(dimethylphosphino)-1,1′-binaphthyl 5a† and 2-(dimethylphosphino)-2′-methoxy-1,1′-binaphthyl 5b.† For each pair, introduction of the heterocycle lowers the energy of both the HOMO and LUMO. Thus we anticipate the phosphiranes to be poorer σ-donors but better π-acceptors than their corresponding unstrained MOP counterpart. Furthermore, the HOMO and LUMO energies for both H-MOP derivatives (S)-2a and 5a were lower than their OMe-MOP counterparts, indicating that one has to consider not just the presence of the strained heterocycle but the backbone as a whole when ascertaining the relative energy levels of these compounds. Calculated bond angles around the phosphorus show a similar degree of pyramidalisation for (S)-2a and (R)-2b (∑(P) 250/251°).
We were keen to study the coordination chemistry of these binaphthyl-based phosphiranes with late transition metals as these are highly relevant for contemporary catalysis, but to date few examples have been reported.8a,10,11 Reaction of two equivalents of (S)-2a or (R)-2b with cis-[Pt(cod)Cl2] resulted in the rapid, quantitative formation of 4a or 4b respectively as shown by HRMS, NMR, and X-ray crystallographic analyses. In the 31P NMR spectra of both complexes the 195Pt satellites accompanying the central peak show large 1JPPt coupling constants (4a: −149.2 ppm, 1JPtP = 4170 Hz; 4b −149.3 ppm, 1JPtP = 4160 Hz). This is a much larger coupling than those observed for the more conventional cis-[Pt(PR3)2Cl2] complexes (1JPtP = 3400–3700 Hz),16 but relates well to that found for the only bisphosphirane platinum dichloride complex previously isolated (1JPtP = 4133 Hz).11b The 10 Hz larger coupling for 4a could be interpreted as a result of the ligand's higher π-acceptor strength in comparison to 4b,6,17 which would also be in agreement with our calculated LUMO energy levels for these phosphirane ligands.†
Crystals suitable for X-ray analysis were grown from concentrated dichloromethane (4a, Fig. 2) or chloroform (4b, Fig. 3) solutions, and both compounds were obtained as solvates. Selected bond lengths and angles are given in the figure captions. Complex 4b has crystallographic two-fold rotational symmetry, with the two phosphirane ligands equivalent, while 4a has no imposed symmetry. The complexes each show a distorted square planar platinum(II) coordination geometry with typical bond lengths and angles about the platinum centre. The high degree of strain in the phosphirane ring is shown by the small C–P–C angles of 50.2(4) and 50.8(4)° for 4a, and 50.9(3)° for 4b. The heterocycles point over the aromatic rings of the second coordinated ligand (closest inter-ligand C⋯C distances: 3.5–3.7 Å). The steric demand of the methoxy group in 4b appears responsible for the increased torsion angle of the binaphthyl backbone, the value of −94.7(8)° being significantly larger than those in 4a of −81.8(10) and −88.5(9)°. These values compare well with the related complex cis-dichlorobis[1-(9-anthracene)phosphirane]platinum(II)11b which exhibits slightly longer Pt–P bond lengths of 2.235(4) and 2.256(4) Å. This anthracenylphosphirane derivative has a slightly higher strain within its heterocyclic ring and is more pyramidalised at phosphorus (C–P–C bond angles: 49.7(8)–49.8(8)°) than 4a or 4b.
![View of the molecular structure of 4a with 50% probability displacement ellipsoids. Hydrogen atoms have been omitted for clarity. Selected bond distances [Å] and angles [°]: Pt–Cl1 2.337(2), Pt–Cl2 2.334(2), Pt–P1 2.212(2), Pt–P2 2.209(2), P1–C41 1.802(3), P1–C42 1.816(8), P1–C1 1.814(7), C41–C42 1.534(11), P2–C43 1.786(8), P2–C44 1.782(7), P2–C21 1.794(8), C43–C44 1.529(11); Cl1–Pt–Cl2 90.52(8), Cl1–Pt–P2 87.06(10), Cl2–Pt–P1 85.40(9), P1–Pt–P2 97.02(8), Pt–P1–C1 118.8(2), C1–P1–C41 107.6(4), C1–P1–C42 111.7(4), C41–P1–C42 50.2(4), Pt–P2–C21 119.1(3), C21–P2–C43 109.8(4), C21–P2–C44 106.4(4), C43–P2–C44 50.8(4); C1–C10–C11–C12 −81.8(10), C21–C30–C31–C32 −88.5(9).](/image/article/2011/CC/c1cc12440d/c1cc12440d-f2.gif) | ||
| Fig. 2 View of the molecular structure of 4a with 50% probability displacement ellipsoids. Hydrogen atoms have been omitted for clarity. Selected bond distances [Å] and angles [°]: Pt–Cl1 2.337(2), Pt–Cl2 2.334(2), Pt–P1 2.212(2), Pt–P2 2.209(2), P1–C41 1.802(3), P1–C42 1.816(8), P1–C1 1.814(7), C41–C42 1.534(11), P2–C43 1.786(8), P2–C44 1.782(7), P2–C21 1.794(8), C43–C44 1.529(11); Cl1–Pt–Cl2 90.52(8), Cl1–Pt–P2 87.06(10), Cl2–Pt–P1 85.40(9), P1–Pt–P2 97.02(8), Pt–P1–C1 118.8(2), C1–P1–C41 107.6(4), C1–P1–C42 111.7(4), C41–P1–C42 50.2(4), Pt–P2–C21 119.1(3), C21–P2–C43 109.8(4), C21–P2–C44 106.4(4), C43–P2–C44 50.8(4); C1–C10–C11–C12 −81.8(10), C21–C30–C31–C32 −88.5(9). | ||
![View of the molecular structure of 4b. Hydrogen atoms have been omitted for clarity. Selected average bond distances [Å] and angles [°]: Pt–Cl 2.338(3), Pt–P 2.204(3), P–C1 1.799(7), P–C2 1.811(7), P–C3 1.802(7), C1–C2 1.550(10); Cl–Pt–Cl′ 90.42(15), Cl–Pt–P 87.28(7), P–Pt–P 96.92(14), Pt–P–C3 115.3(2), C1–P–C3 110.6(3), C2–P–C3 106.0(3), C1–P–C2 50.9(3); C3–C12–C13–C22 −94.7(8). A prime indicates a symmetry-equivalent atom.](/image/article/2011/CC/c1cc12440d/c1cc12440d-f3.gif) | ||
| Fig. 3 View of the molecular structure of 4b. Hydrogen atoms have been omitted for clarity. Selected average bond distances [Å] and angles [°]: Pt–Cl 2.338(3), Pt–P 2.204(3), P–C1 1.799(7), P–C2 1.811(7), P–C3 1.802(7), C1–C2 1.550(10); Cl–Pt–Cl′ 90.42(15), Cl–Pt–P 87.28(7), P–Pt–P 96.92(14), Pt–P–C3 115.3(2), C1–P–C3 110.6(3), C2–P–C3 106.0(3), C1–P–C2 50.9(3); C3–C12–C13–C22 −94.7(8). A prime indicates a symmetry-equivalent atom. | ||
Having established that the thermal and air-stability of our phosphiranes does not infringe on their ability to coordinate to a late transition metal, we sought to extend the research to include a pilot study of the potential of these ligands for asymmetric catalysis. As a benchmark, the asymmetric induction capabilities of (S)-2a and (R)-2b were investigated in the catalytic hydrosilylation of styrene (Scheme 3).18 The reaction is required to be both highly regio- and enantioselective; oxidation of the initially formed trichlorosilane proceeds with retention of configuration and chiral 1-phenylethanol is produced (Scheme 3). Our catalysts were prepared in situ by reacting the chiral phosphirane ligand with [PdCl(η3-C3H5)]2.
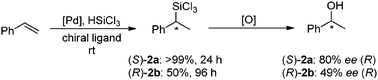 | ||
| Scheme 3 The asymmetric hydrosilylation of styrene gives the chiral trichlorosilane, subsequent oxidation of which gives 1-phenylethanol. | ||
The results obtained with the two ligands differ significantly. The hydrogen substituent in the 2′-position of the binaphthyl backbone seems to promote the activity as well as the enantioselectivity of the catalyst. A similar trend was found for the related parent MOP ligands when used in this transformation.19 The full conversion obtained with (S)-2a coupled with attaining an enantiomeric excess of 80% indicates the potential applications of this neglected class of ligand. To the best of our knowledge this is the first time that phosphiranes with the chirality located solely on the ligand backbone have been utilised in asymmetric catalysis.10 Note these reaction conditions are not optimised and the results represent only a first glance at the catalytic potential of these ligands.
We thank the European Union (Erasmus Undergraduate Exchange for I.M.-B.) and Newcastle University for funding. We also thank the EPSRC for a Career Acceleration Fellowship (L.J.H.), a Studentship (A.F.), an equipment grant (W.C.), and its National Mass Spectrometry Service Centre, Swansea, UK.
Notes and references
- Reviews: (a) F. Mathey, Chem. Rev., 1990, 90, 997 CrossRef CAS; (b) L. D. Quin, in A Guide to Organophosphorus Chemistry, ed. L. D. Quin, John Wiley Sons, New York, 2000, pp. 234-241 Search PubMed; (c) F. Mathey and M. Regitz, in Phosphorus–Carbon Heterocyclic Chemistry: The Rise of a New Domain, ed. F. Mathey, Elsevier Science, Amsterdam, 2001, pp. 17–55 Search PubMed.
- Phosphirane decomposes completely within 24 h at 25 °C: R. I. Wagner, L. D. Freeman, H. Goldwhite and D. G. Rowsell, J. Am. Chem. Soc., 1967, 89, 1102 CrossRef CAS.
- (a) Y. B. Kang, M. Pabel, A. C. Willis and S. B. Wild, J. Chem. Soc., Chem. Commun., 1994, 475 RSC; (b) D. C. R. Hockless, Y. B. Kang, M. A. McDonald, M. Pabel, A. C. Willis and S. B. Wild, Organometallics, 1996, 15, 1301 CrossRef CAS.
- X. Li, K. D. Robinson and P. P. Gaspar, J. Org. Chem., 1996, 61, 7702 CrossRef CAS.
- M. Baudler and J. Germeshausen, Chem. Ber., 1985, 118, 4285 CrossRef CAS.
- T. Oshikawa and M. Yamashita, Synthesis, 1985, 290 CrossRef CAS.
- (a) J. Liedtke, S. Loss, G. Alcaraz, V. Gramlich and H. Grützmacher, Angew. Chem., 1999, 111, 1724 ( Angew. Chem., Int. Ed. , 1999 , 38 , 1623 ) CrossRef; (b) J. Liedtke, S. Loss, C. Widauer and H. Grützmacher, Tetrahedron, 2000, 56, 143 CrossRef CAS; (c) J. Liedtke, H. Rüegger, S. Loss and H. Grützmacher, Angew. Chem., 2000, 112, 3596 ( Angew. Chem., Int. Ed. , 2000 , 39 , 2478 ) CrossRef; (d) C. Laporte, G. Frison, H. Grützmacher, A. C. Hillier, W. Sommer and S. P. Nolan, Organometallics, 2003, 22, 2202 CrossRef CAS.
- (a) S. Maurer, C. Burkhart and G. Maas, Eur. J. Org. Chem., 2010, 2504 CrossRef CAS; (b) T. Jikyo and G. Maas, Chem. Commun., 2003, 2794 RSC.
- R. M. Hiney, L. J. Higham, H. Müller-Bunz and D. G. Gilheany, Angew. Chem., 2006, 118, 7406 ( Angew. Chem., Int. Ed. , 2006 , 45 , 7248 ) CrossRef.
- Phosphiranes with chirality on the heterocycle have been used in asymmetric hydrogenations: A. Marinetti, F. Mathey and L. Ricard, Organometallics, 1993, 12, 1207 CrossRef CAS and references therein.
- (a) N. Mézailles, P. E. Fanwick and C. P. Kubiak, Organometallics, 1997, 16, 1526 CrossRef; (b) F. Yang, P. E. Fanwick and C. P. Kubiak, Organometallics, 1999, 18, 4222 CrossRef CAS.
- H. R. Hudson, in The Chemistry of Organophosphorus Compounds, ed. F. R. Hartley, Wiley, New York, 1990, vol. 1, pp. 438–439 and references therein Search PubMed.
- B. Stewart, A. Harriman and L. J. Higham, submitted to Organometallics Search PubMed , om-2011-00070a.
- P. P. Gaspar, H. Qian, A. M. Beatty, D. André d'Avignon, J. L.-F. Kao, J. C. Watt and N. P. Rath, Tetrahedron, 2000, 56, 105 CrossRef CAS.
- (a) D. C. R. Hockless, M. A. McDonald, M. Pabel and S. B. Wild, J. Chem. Soc., Chem. Commun., 1995, 257 RSC; (b) D. C. R. Hockless, M. A. McDonald, M. Pabel and S. B. Wild, J. Organomet. Chem., 1997, 529, 189 CrossRef CAS.
- P. S. Pregosin, Annu. Rep. NMR Spectrosc., 1986, 17, 285 CrossRef.
- C. M. Haar, S. P. Nolan, W. J. Marshal, K. G. Moloy, A. Prock and W. P. Giering, Organometallics, 1999, 18, 474 CrossRef CAS.
- See e.g. J. W. Han and T. Hayashi, Tetrahedron: Asymmetry, 2010, 21, 2193 CrossRef CAS and references therein.
- K. Kitayama, Y. Uozumi and T. Hayashi, J. Chem. Soc., Chem. Commun., 1995, 1533 RSC ; at 0 °C (S)-H-MOP gave full conversion in 12 h with 93% ee, (R)-MOP required 24 h for full conversion (14% ee).
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and crystallographic data. CCDC 816636 (4a) and 816637 (4b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1cc12440d |
| This journal is © The Royal Society of Chemistry 2011 |