Stability of small mesoporous silica nanoparticles in biological media†‡
Yu-Shen
Lin
,
Nardine
Abadeer
and
Christy L.
Haynes
*
Department of Chemistry, University of Minnesota, 207 Pleasant Street SE, Minneapolis, Minnesota, 55455, USA. Web: http://www.chem.umn.edu/groups/haynesE-mail: chaynes@umn.edu; Fax: +1 612-626-7541; Tel: +1 612-626-1096
First published on 16th November 2010
Abstract
In this work, sub-50 nm pegylated mesoporous silica nanoparticles prepared with hydrothermal treatment are shown to have long-term stability in various media at both room and physiological temperature. Compared to bare mesoporous silica nanoparticles, the highly pegylated mesoporous silica nanoparticles show significantly improved biocompatibility and decreased macrophage uptake, making these nanoparticles viable for in vivo stealth drug delivery applications.
Mesoporous silica (MS) nanoparticles (NPs) were first imagined as a vehicle for drug delivery by Lin et al.;1 since then, many researchers have begun to explore the potential of these NPs for various biomedical applications such as intracellular labeling,2 biomolecule delivery,3 targeting,4 and diagnostic imaging.5 Although much published work has successfully shown the in vitro therapeutic promise of MS NPs, very few in vivo experiments can be found in the literature due to the large size (>150 nm diameter) and low stability of MS NPs in biological media, resulting in rapid unintentional uptake by the reticuloendothelial system (RES).6–8 These two critical issues must be solved if MS NPs are ever going to fulfill their potential for in vivo biological use. Although synthesis of fine bare MS NPs (<50 nm) has been reported by several groups recently,9–11 particle dispersity has been difficult to maintain, and aggregation occurs either during synthesis or surfactant removal.10 Removal of the surfactant template from MS NPs is the key step during the synthetic procedure that determines retention of the initial dispersity of MS NPs, especially for small MS NPs (i.e. diameters less than 50 nm). Very recent work published by Kuroda et al. showed that using a dialysis process to remove surfactant can prevent aggregation of small MS NPs and maintain their hydrodynamic size.11 Another popular strategy to maintain NP dispersity is via surface passivation with polyethylene glycol (PEG); PEG has been known to prevent protein adsorption (opsonization) on nanoparticle surfaces, enhance circulation time, and reduce nonspecific RES uptake.12,13 Though the pegylation of MS NPs has been shown to reduce their hemolytic activity,14 serum binding,15a and short-term degradation in simulated body fluid,15b there is no precedent work examining the long-term stability of these pegylated MS NPs in various biological media such as phosphate buffered saline (PBS) or cell culture media at 37 °C. This paucity of work, on a feature critical to the application of these materials, reveals that synthesizing MS NPs with long-term colloidal stability is still a challenge in this field.
Another major hurdle to biological application of MS NPs is that the hydrodynamic size and aggregation state of MS NPs, as determined by dynamic light scattering (DLS), is only examined in deionized (D.I.) water9 or organic solvents.11 In most cases, biomedical MS NPs will be suspended in or experience highly salted solutions or serum-containing media, conditions likely to have a large influence on the hydrodynamic diameter. In addition, the short-term stability (hours) of MS NPs may be different from their long-term stability (days). To directly address the aforementioned gaps in MS NP research, this communication details the examination of nanoparticle stability in various biological media over a long period of time (10 days) at 37 °C to mimic realistic biological conditions. We aim to synthesize MS NPs with hydrodynamic diameter less than 100 nm and long-term stability in biological media at physiological temperature.
Herein, we prepared small MS NPs (<50 nm) with well-ordered pore structure based on our previously published protocol.14a The template of the as-synthesized bare MS NPs with 42 nm diameter (MS42) was removed by two different methods, centrifugation14b or dialysis,9 and accordingly, are designated as MS42-c and MS42-d, respectively (Scheme 1). In the preparation of pegylated MS NPs, a short polyethylene glycol silane (PEG-silane) was added to functionalize the exterior of the MS42 NPs using a delay modification method.16 In some cases, a hydrothermal treatment was introduced (to promote NP stability) before pegylated MS NP template removal was accomplished by centrifugation; this product is designated as MS42@PEG-hy-c (Scheme 1). Pegylated MS NPs without hydrothermal treatment, for comparison, are named as MS42@PEG-c. All details of synthesis and characterization are described in the ESI.‡
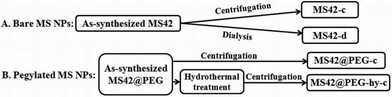 | ||
| Scheme 1 Preparation flowchart of bare and pegylated MS NPs: MS42-c, MS42-d, MS42@PEG-c, and MS42@PEG-hy-c. | ||
Transmission electron microscopy (TEM) images of MS42-c, MS-42-d, MS42@PEG-c, and MS42@PEG-hy-c showed that all particles had similar diameters of approximately 40 nm (Fig. 1a,b and Fig. S1 (ESI‡)). The TEM images clearly reveal that the pore structure inside hydrothermally treated NP was not as ordered as the other three MS NPs. Compared to MS42-d NPs, MS42@PEG-hy-c NPs also had a lower number of X-ray diffraction (XRD) peaks (Fig. 1c) and a shift in the N2 adsorption–desorption isotherm capillary condensation to lower pressure (Fig. 1d). Additionally, the total surface area of MS42@PEG-hy-c greatly decreased from 1131 (MS42-d) to 731 m2 g−1 and pore size changed from 2.60 nm to 1.94 nm (see Table S2 in ESI‡). These data support that pore order was altered by PEG-silane surface modification and/or incorporation into MS NP framework during the hydrothermal treatment. By comparing the difference in surface area (from 943 to 731 m2 g−1) and pore size (from 2.51 to 1.94 nm) between MS42@PEG-c and MS42@PEG-hy-c (see Table S2 in ESI‡), confirms that PEG-silane modifies the internal pore and incorporates into the pore wall of MS NPs during the hydrothermal pore restructuring process.
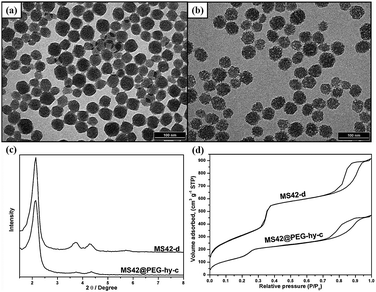 | ||
| Fig. 1 TEM images of surfactant-free (a) MS42-d and (b) MS42@PEG-hy-c NPs. (c) XRD patterns of surfactant-free MS42-d and MS42@PEG-hy-c NPs. (d) N2 adsorption–desorption isotherms of surfactant-free MS42-d and MS42@PEG-hy-c NPs. | ||
The hydrodynamic diameter of the as-synthesized and extracted MS NPs in D.I. water at room temperature (RT) is summarized in Table S1, ESI.‡ Except for the bare MS NPs purified using centrifugation, MS42-c, the hydrodynamic size of surfactant-free bare and pegylated MS NPs was similar to their as-synthesized counterparts (Fig. 2a and Table S1 in ESI‡), showing that aggregation occurs during the high-speed centrifugation and redispersion of NPs viaultrasonication. Compared to MS42-d and MS42@PEG-hy-c NPs, the MS42-c NPs aggregated easily and precipitated after 30 minutes aging in PBS (see Fig. S2 in ESI‡). Fig. 2b shows a slight increase in hydrodynamic diameter for MS42-d NPs when they were dispersed in PBS and a significant size change upon dispersion into Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) (Fig. 2c). The size change of the MS42-d NPs in both PBS and DMEM + 10%FBS is likely due to charge neutralization of surface silanol groups by ionic species and rapid adsorption of proteins on the nanoparticle surface. Additionally, further examination of the long-term stability of MS42-d and MS42@PEG-hy-c in D.I. water, PBS, and DMEM + 10% FBS at RT and 37 °C was performed (Fig. 2d and e). Both nanoparticles were very stable in D.I. water, but MS42-d aggregated in highly salted media, and the temperature effect is profound for the bare MS NPs, with higher incubation temperature causing more extensive aggregation. For MS42@PEG-hy-c NPs, no significant size change was observed after even 10 days of aging in biological media at either RT or 37 °C, except for the PBS aging at 37 °C. Even though the hydrodynamic size of MS42@PEG-hy-c NPs gradually increased after 6-day PBS aging at 37 °C, the aggregates are still small (less than 100 nm). No visible particle precipitation was observed for these NPs after 10-day incubation in D.I. water, PBS, or DMEM + 10%FBS at 37 °C (see inset in Fig. 2e). The stability comparison between MS42@PEG-c and MS42@PEG-hy-c NPs in PBS further showed that the hydrothermal treatment significantly improved the long-term stability of MS42@PEG-hy-c NPs (see Fig. S3 in ESI‡). This is likely due to a greater number of PEG groups either on the outer or inner surface of the nanoparticles as well as more complete silica condensation between silanol groups following hydrothermal treatment.
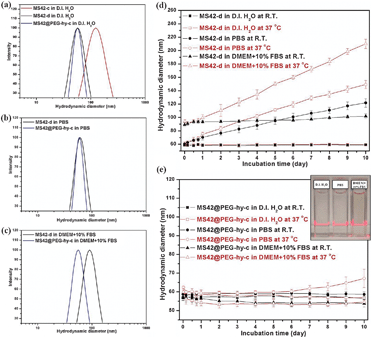 | ||
| Fig. 2 Hydrodynamic size distribution of 1 mg mL−1 MS-42-c, MS42-d, and MS42@PEG-hy-c NPs measured by DLS at RT in various media: (a) D.I. H2O, (b) PBS, and (c) DMEM + 10%FBS. Long-term colloidal stability of (d) MS42-d and (e) MS42@PEG-hy-c NPs in various media at RT and 37 °C. Data represent mean ± SD from three independent experiments. Inset: a photograph of MS42@PEG-hy-c colloidal solutions after 10-day aging in D.I. H2O, PBS, and DMEM + 10%FBS at 37 °C. | ||
Fig. 3a shows the amount of free silicic acid, a clear indicator of NP degradation, from MS42-d and MS42@PEG-hy-c after 10-days in D.I. water and PBS incubation at RT and 37 °C, as quantified by a blue silicomolybdic assay.17 The degraded amounts from MS42-d were greater than MS42@PEG-hy-c after both 10 days in D.I. water and PBS at RT and 37 °C. Additionally, the aging temperature influenced the amount of dissolved silica. Higher temperature led to more silica dissolution in both D.I. water and PBS aging conditions. We also examined the XRD patterns of MS42-d and MS42@PEG-hy-c after 10-day aging in D.I. water and PBS at RT and 37 °C. In the case of D.I. water aging, the XRD patterns of aged MS42-d and MS42@PEG-hy-c NPs were almost the same as the unaged particles (see Fig. S4 in ESI‡). In contrast, the pore structure of MS42-d and MS42@PEG-hy-c collapsed after 10-day PBS aging (Fig. 3b and c). In both particles, the extent of pore collapse was more extensive at 37 °C than at RT. While the (100) peak of MS42-d NPs after 10-day PBS aging at 37 °C is almost undetectable, it is still apparent in the aged MS42@PEG-hy-c NPs. The evolution of porosity of MS42@PEG-hy-c is also examined by XRD and N2 physisorption measurements. The XRD pattern of MS42@PEG-hy-c changed from three distinct peaks to one peak after 1-day PBS aging at 37 °C (see Fig. S5 in ESI‡). The pore structure is then almost unchanged even after 10-day PBS aging at 37 °C. Additionally, the percent retention of surface area of MS42-d and MS42@PEG-hy-c were 44% and 59%, respectively (see Table S2 in ESI‡). These data indicate the enhanced pore retention achieved by pegylation of MS42 NPs during hydrothermal treatment. The comparison of dissolved silica amounts and the pore structure after 10-day PBS aging of MS42-d and MS42@PEG-hy-c NPs further demonstrates that the PEG modification and hydrothermal treatment of the MS NPs reduces the extent of MS NP degradation within biological media, yielding a stable nanoparticle with significant promise for in vivo applications.
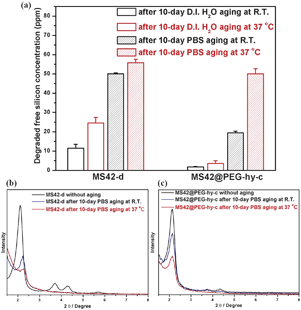 | ||
| Fig. 3 (a) Degraded free silicon concentration from 1 mg mL−1MS42-d and MS42@PEG-hy-c suspensions after 10-day aging in D.I. H2O and PBS at RT and 37 °C. Data represent mean ± SD from three independent experiments. XRD patterns of (b) MS42-d and (c) MS42@PEG-hy-c NPs after 10-day PBS aging at RT. and 37 °C. | ||
To evaluate the biocompatibility of these stable MS42@PEG-hy-c NPs, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) and hemolysis assays were used. The results in Fig. 4a and b show that human endothelial cells and red blood cells are not compromised even after 24-h exposure at a relatively high concentration (1000 μg mL−1). Additionally, the stealth property of MS42@PEG-hy-c NPs was studied by monitoring macrophage uptake. Fig. 4c showed that MS42@PEG-hy-c NPs exhibited 70% reduction in the amount of macrophage uptake compared to bare MS NPs, MS42-d. The uptake percentage of MS42@PEG-hy-c NPs (200 μg mL−1) by macrophages after 24-hour exposure was only 0.5%, further confirming that pegylated MS NPs with hydrothermal treatment yields greatly reduced protein adsorption, resulting in a resistance to nonspecific uptake by macrophage cells.
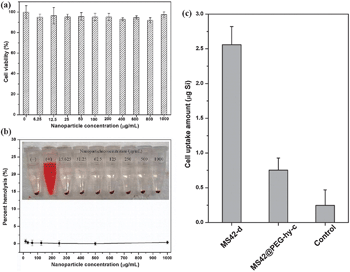 | ||
| Fig. 4 (a) Cell viability of human endothelial cells after exposure to different concentrations of MS42@PEG-hy-c NPs. Data represent mean ± SD from four replicates. (b) Percent hemolysis of RBCs incubated with different concentrations of MS42@PEG-hy-c NPs for 3 hours at 37 °C; inset is the photograph of RBCs after NP exposure. Data represent mean ± SD from four replicates. (c) Uptake amounts of silicon by Raw 264.7 macrophages after 24-hour exposure of 200 μg mL−1 of MS42-d and MS42@PEG-hy-c NPs. Data represent mean ± SD from three independent experiments with four replicates. | ||
This work was funded by NSF (CHE-0645041). Y.-S. L. and N. A. acknowledge financial support from a Taiwan Merit Scholarship and Heisig/Gleysteen Program, respectively. TEM and XRD characterization were carried out in the College of Science and Engineering Characterization Facility, which receives partial support from NSF through NNIN.
Notes and references
- C.-Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451 CrossRef CAS.
- Y.-S. Lin, C.-P. Tsai, H.-Y. Huang, C.-T. Kuo, Y. Hung, D.-M. Huang, Y.-C. Chen and C.-Y. Mou, Chem. Mater., 2005, 17, 4570 CrossRef CAS.
- I. I. Slowing, B. G. Trewyn and V. S.-Y. Lin, J. Am. Chem. Soc., 2007, 129, 8845 CrossRef CAS.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Lindén, ACS Nano, 2009, 3, 197 CrossRef CAS.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438 CrossRef CAS.
- S.-H. Wu, Y.-S. Lin, Y. Hung, Y.-H. Chou, Y.-H. Hsu, C. Chang and C.-Y. Mou, ChemBioChem, 2008, 9, 53 CrossRef CAS.
- K. M. L. Taylor, J. S. Kim, W. J. Rieter, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2008, 130, 2154 CrossRef CAS.
- C.-H. Lee, S.-H. Cheng, Y.-J. Wang, Y.-C. Chen, N.-T. Chen, J. Souris, C.-T. Chen, C.-Y. Mou, C.-S. Yang and L.-W. Lo, Adv. Funct. Mater., 2009, 19, 215 CrossRef CAS.
- J. Kobler, K. Möller and T. Bein, ACS Nano, 2008, 2, 791 CrossRef CAS.
- F. Lu, S.-H. Wu, Y. Hung and C.-Y. Mou, Small, 2009, 5, 1408 CrossRef CAS.
- C. Urata, Y. Aoyama, A. Tonegawa, Y. Yamachi and K. Kuroda, Chem. Commun., 2009, 5094 RSC.
- C. Fang, N. Bhattarai, C. Sun and M. Zhang, Small, 2009, 5, 1637 CrossRef CAS.
- I. M. Rio-Echevarria, F. Selvestrel, D. Segat, G. Guarino, R. Tavano, V. Causin, E. Reddi, E. Papini and F. Mancin, J. Mater. Chem., 2010, 20, 2780 RSC.
- (a) Y.-S. Lin and C. L. Haynes, J. Am. Chem. Soc., 2010, 132, 4834 CrossRef CAS; (b) Y.-S. Lin and C. L. Haynes, Chem. Mater., 2009, 21, 3979 CrossRef CAS.
- (a) Q. He, J. Zhang, J. Shi, Z. Zhu, L. Zhang, W. Bu, L. Guo and Y. Chen, Biomaterials, 2010, 31, 1085 CrossRef CAS; (b) V. Cauda, A. Schlossbauer and T. Bein, Microporous Mesoporous Mater., 2010, 132, 60 CrossRef CAS.
- (a) J. Kecht, A. Schlossbauer and T. Bein, Chem. Mater., 2008, 20, 7207 CrossRef CAS; (b) V. Cauda, A. Schlossbauer, J. Kecht, A. Zürner and T. Bein, J. Am. Chem. Soc., 2009, 131, 11361 CrossRef CAS.
- T. Coradin, D. Eglin and J. Livage, Spectrosc. – Int. J, 2004, 18, 567 Search PubMed.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, tables, and additional figures. See DOI: 10.1039/c0cc02923h |
| This journal is © The Royal Society of Chemistry 2011 |