A general route for the synthesis of functional, protein-based hydrogel microspheres using tailored protein charge†‡
William J.
King
a,
Michael W.
Toepke
a and
William L.
Murphy
*abc
aThe University of Wisconsin, Department of Biomedical Engineering, 1550 Engineering Drive, Madison, WI, USA. E-mail: wlmurphy@wisc.edu; Fax: 6082659239; Tel: 6082654031
bThe University of Wisconsin, Department of Pharmacology, 1300 University Dr, Madison, WI, USA
cThe University of Wisconsin Department of Orthopedics and Rehabilitation, 777 Highland Ave., Madison, WI, USA
First published on 4th November 2010
Abstract
pH-induced protein aggregation facilitated the formation of nucleation centers for the chain growth polymerization of hydrogel microspheres.
The development of protein-based materials has emerged as a significant area of chemical research, as incorporated proteins have imbued materials with unique functions. A potentially important approach has involved including proteins in hydrogel microparticles, which are hydrophilic, geometrically tailorable, and useful in diverse applications. In one relevant example, Lee et al. used photolithography to form poly(ethylene glycol) (PEG)-based hydrogel microparticles that contained the proteins glucose oxidase, peroxidase, or alkaline phosphatase. They used differences in microparticle geometry to simultaneously characterize the enzymatic reaction rates of the encapsulated proteins.1 In this approach and others, it is clear that control over microparticle geometry and protein activity is critical for the successful application of these functional materials.
We hypothesized that hydrogel microspheres with controllable properties could be formed via a surfactant-free polymerization process. In our proposed process, macromolecules form polymerizable homo-aggregates dispersed in a continuous phase. The mechanisms that control protein aggregation have been well characterized, and therefore protein aggregation could serve as a broadly applicable approach to form nucleation centers for microsphere polymerization. The extent of protein aggregation has been shown to be dependent upon pH, salt concentration, salt type, temperature, and protein concentration. However, a protein's minimum solubility normally occurs at a pH around its isoelectric point (pI), which has been attributed to the lack of electrostatic repulsion.2 For example, lysozyme's pI has been measured to be 10.5, and its minimum solubility was measured to be at pH 10.3 Therefore, we reasoned that varying pH around a protein's pI could be used to modulate formation of protein aggregates, which could serve as nucleation centers in suspension polymerizations.
In this communication we describe a generalizable approach to form hydrogel microspheres based on pH-induced protein aggregation. Specifically, we hypothesized that varying the pH around a protein's pI would modulate the degree of protein aggregation and the size of the resulting microspheres (Fig. 1). We herein refer to this approach as the pH-induced protein aggregation or the “PIPA” approach.
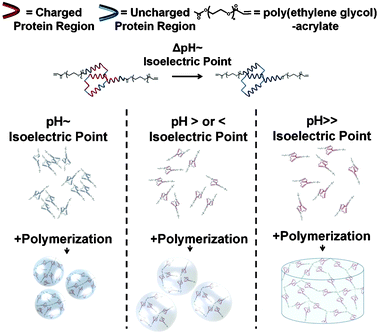 | ||
| Fig. 1 Schematic representation of the hypothesized effect of pH on protein aggregation and hydrogel microsphere formation. The protein-polymer conjugates form dense aggregates and small microspheres when pH ∼ protein's pI (left). The conjugates form less dense aggregates and form larger microspheres when pH is > or < protein's pI (middle). When pH ≫ protein's pI, the protein-polymer conjugates do not aggregate and form a macroscopic hydrogel disk (right). | ||
To demonstrate the feasability of our PIPA approach, we formed a model protein-polymer conjugate by reacting mutant calmodulin CaM(T34C, T110C) with PEG-diacrylate (Mn = 3400 Da) to render “PEG-CaM-PEG” conjugates with polymerizable acrylate end groups.4–7 Then, we prepared 10 mM PEG-CaM-PEG solutions with varying pH around CaM's isoeletric point, 4.2, using citric acid buffer (20 mM Citric Acid, 150 mM NaCl, and 10 mM CaCl2) or MES buffer (20 mM MES, 150 mM NaCl, and 10 mM CaCl2). 0.05% Irgacure 2959 (Ciba, Basel, Switzerland) was included as a polymerization initiator. The combination of protein-polymer conjugates, polymerization initiator, and buffer will herein be referred to as a “prepolymer solution.”
To validate the broad based applicability of this approach we also formed another protein-polymer conjugate by reacting the putative bacterial hyaluronidase (Hase) TDE0471 [Hase(21C, 221C, 516C)] (a kind gift from Professor J. Christopher Fenno, University of Michigan) with PEG-diacrylate (Mn = 3400 Da) to form “Hase-PEG3” conjugates (ESI,† Fig. 1). 5 μl prepolymer solutions were cast between two glass slides with a 1 mm spacer, and exposed to UV light (4 mW/cm2) for 0.5 min to initiate polymerization. Bright field micrographs of the resulting swollen microspheres were acquired using an inverted microscope (Nikon, Melville, NY, USA). The diameters of >500 microspheres per experimental condition were measured using ImageJ software (Freeware, NIH, Bethesda, MD).
PEG-CaM-PEG conjugates prepared at pH conditions close to CaM's pI (4.2) formed microspheres upon polymerization. Microspheres formed at pH 3.2, pH 4.2, and pH 5.2 had average diameters of 2.1 ± 0.3 μm, 2.2 ± 0.4 μm, and 2.2 ± 0.3 μm, respectively (Fig. 2, confocal microgaphs in ESI,† Fig. 2, and Supplementary Movie 1). Also, microspheres formed at a 20× greater scale formed with an average diameter of 2.2 ± 0.9 μm which was not statistically different from those formed at normal scale (ANOVA, p < 0.05) (ESI,† Fig. 3). It is noteworthy that these average diameters and size distributions were nearly an order of magnitude smaller and much less variable in size than the 38.1 ± 16.1 μm average diameter of PEG-CaM-PEG microspheres previously formed using a water-in-oil suspension polymerization.5 This difference in size distributions between microspheres formed here using the PIPA approach and previously formed using a suspension polymerization is in agreement with previously reported trends across a broad array of polymeric microspheres.8
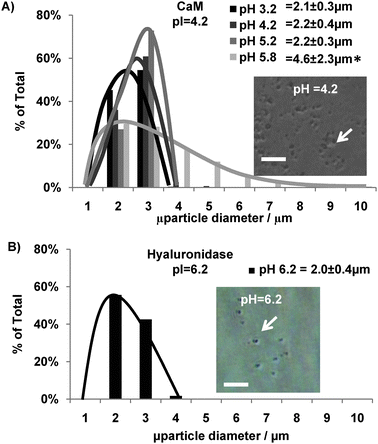 | ||
| Fig. 2 Effect of pH on hydrogel microsphere size distribution. (A) Size distribution of PEG-CaM-PEG microspheres prepared at different pH conditions with an inlayed representative micrograph of microspheres formed at pH 4.2. * denotes a significant difference in microsphere diameter compared to microspheres formed at pH 4.2. ANOVA p < 0.05. (B) Size distribution of Hase-PEG3 microspheres prepared at Hase's pI with an inlayed representative micrograph of microspheres formed at pH 6.2. Scalebar for A–B denotes 30 μm. | ||
Prepolymer solutions formed at pH conditions further away from CaM's pI formed either larger microspheres or failed to form microspheres and instead formed a singular macroscopic hydrogel disk. For example, microspheres formed at pH 5.8 had an average diameter of 4.6 ± 2.3 μm which was significantly larger than the microspheres formed at pH 4.2 and had a larger size distribution (Fig. 2). When the pH was shifted even further away from CaM's pI, to pH 6.3, no microspheres formed after 30 s of polymerization. However, after 5 min of polymerization, a singular 5 μl macroscopic hydrogel disk formed between the glass slides due to CaM's highly charged state at pH 6.3 (ESI,† Fig. 4). This result suggested that varying the prepolymer solution pH further away from CaM's pI increased CaM's charge, induced electostatic repulsion between PEG-CaM-PEG molecules, and inhibited microsphere formation.
The PIPA approach also formed hydrogel microspheres from another protein-polymer conjugate, demonstrating a potential for broad applicability. The Hase protein used in this study had a different molecular weight (60.7 kDa), pI (6.2), and number of reactive cysteines (3) compared to the mutant CaM (16.7 kDa, pI 4.2, and 2 reactive cysteines). Despite these physiochemical differences, the PIPA approach enabled the formation of hyaluronidase-PEG3 microspheres with an average diameter and size distribution similar to PEG-CaM-PEG microspheres (Fig. 2 and Supplementary Fig. 1). These results demonstrated that the PIPA approach could facilitate the incorporation of proteins with distinct physiochemical properties into hydrogel microspheres. The ability to incorporate dissimilar proteins into hydrogel microspheres could enable the development of multifunctional materials in future studies.
The well-characterized surfactant-free precipitation polymerization process described previously9 is similar to the PIPA approach used in this study, and therefore could provide insight into the mechanism controlling the formation of PEG-CaM-PEG microspheres here. For example, during previous precipitation polymerizations, synthetic polymer aggregation was necessary to induce microsphere formation. In one illustrative study, Pelton and Chibante formed poly(N-isopropylacrylamide) (poly(NIPAAM))-based microspheres when the prepolymer solution was polymerized above 60 °C, which is well above poly(NIPAAM)'s 32 °C lower critical solution temperature. However, when the prepolymer solution temperature was decreased to 55 °C no microspheres formed.10 This adaptability may be beneficial in the formation of future hydrogel microspheres, as determining a protein's charge and solubility at different pHs is difficult due to the complex intermolecular interactions involved in protein folding.11
We next determined whether CaM retained its function after incorporation into the hydrogel network. We have used several approaches to demonstrate that CaM's nanometre-scale conformational change has translated to macroscopic volume changes when it was incorporated as a functional unit in hydrogel networks. First, hydrogels formed with another mutant CaM(Q41C,K75C), in which the distance between cysteines underwent a smaller magnitude distance change, underwent smaller magnitude volume changes. Also, arginine (which has the same charge as the CaM-specific ligand trifluoperazine (TFP)), bovine serum, and a scrambled version of a CaM peptide ligand did not induce CaM-based hydrogel volume changes. Furthermore, CaM's binding affinity for TFP was similar in hydrogel networks and in solution.4–7,12 Together, these results indicated that CaM maintains its function in hydrogel networks and its nanometre scale conformational change has translated to macroscopic hydrogel volume changes, and we therefore used microsphere volume changes as an indicator of CaM function in the current study.
In this study, each population of PEG-CaM-PEG microspheres underwent ligand-induced volume changes, which indicated that CaM maintained its functionality after incorporation into hydrogel microspheres (Fig. 3). The magnitude of the volume changes observed for microspheres formed at pH 4.2 (42 ± 30% of initial volume) was similar to the magnitude of the volume changes observed in previous studies for PEG-CaM-PEG hydrogel microspheres formed using a water-in-oil supension polymerization (53 ± 3% of initial volume)5 and macroscopic hydrogel disks (10–80% of initial volume, depending on initial PEG-CaM-PEG concentration).4 Together, these results suggested that CaM's biological activity was not adversely affected when incorporated into microspheres using the PIPA approach.
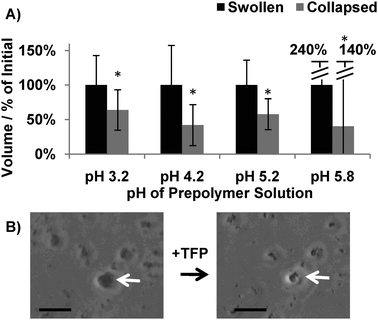 | ||
| Fig. 3 Effect of the PIPA microsphere processing approach on protein activity. (A) Quantification of PEG-CaM-PEG microspheres' ligand-induced volume changes. * denotes a significant difference compared to swollen microspheres. ANOVA p < 0.05 (n > 500 microspheres). (B) Micrographs of microspheres that were formed at pH 5.8 before and after exposure to a CaM-specific ligand, TFP. Scalebar denotes 10 μm. | ||
The microsphere synthesis approach described here may be broadly applicable. The pIs of thousands of proteins are known, can be determined experimentally, or predicted using online databases.13,14 Also, the pI of peptides or proteins could be tailored using solid phase peptide synthesis or recombinant protein engineering techniques.15 Furthermore, there are numerous chemistries available to covalently conjugate polymers to proteins which could facilitate further control over the pI of polymer-protein-polymer conjugates.16 Other advantages of this PIPA approach include that it requires no organic phase, surfactants, or purification steps. Together, the unique attributes of this approach could enable new chemical applications for protein-based hydrogel microspheres formed over a well controlled size range with active incorporated proteins.
The authors are grateful for support from the National Science Foundation (Materials Research Science and Engineering Center seed grant to W.L.M., Graduate Research Fellowship to W.J.K., and CAREER award #0745563 to W.L.M.) and the National Institutes of Health (Training Grant T32 DC009401 to MWT).
References
- W. Lee, D. Choi, J. H. Kim and W. G. Koh, Biomed Microdevices, 2008, 10, 813–822 CrossRef CAS.
- O. D. Velev, E. W. Kaler and A. M. Lenhoff, Biophys. J., 1998, 75, 2682–2697 CAS.
- Y. C. Shih, J. M. Prausnitz and H. W. Blanch, Biotechnol. Bioeng., 1992, 40, 1155–1164 CrossRef.
- W. King, J. Mohammed and W. Murphy, Soft Matter, 2009, 5, 2399–2406 RSC.
- W. J. King, J. N. Pytel, K. Ng and W. L. Murphy, Macromol Biosci, 2010, 10, 580–584 CrossRef CAS.
- Z. J. Sui, W. J. King and W. L. Murphy, Adv. Mater., 2007, 19, 3377–3380 CrossRef CAS.
- Z. J. Sui, W. J. King and W. L. Murphy, Adv Funct Mater, 2008, 18, 1824–1831 CrossRef CAS.
- H. Kawaguchi, Prog. Polym. Sci., 2000, 25, 1171–1210 CrossRef CAS.
- D. L. Elbert, Acta Biomater., 2010 DOI:10.1016/j.actbio.2010.07.028.
- R. H. Pelton and P. Chibante, Colloids and Surf., 1986, 20, 247–256 CrossRef CAS.
- D. Bashford and M. Karplus, Biochemistry, 1990, 29, 10219–10225 CrossRef CAS.
- W. L. Murphy, W. S. Dillmore, J. Modica and M. Mrksich, Angew. Chem. Int. Ed. Engl., 2007, 46, 3066–3069 CrossRef CAS.
- H. Berman, K. Henrick and H. Nakamura, Nat. Struct. Biol., 2003, 10, 980–980 CrossRef CAS.
- B. J. Cargile, J. L. Bundy, T. W. Freeman and J. L. Stephenson, J. Proteome Res., 2004, 3, 112–119 CrossRef CAS.
- G. B. Fields and R. L. Noble, Int. J. Pept. Protein Res., 1990, 35, 161–214 CAS.
- M. A. Gauthier and H. A. Klok, Chem. Commun., 2008, 2591–2611 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Further figures and movie. See DOI: 10.1039/c0cc02716b |
| ‡ This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| This journal is © The Royal Society of Chemistry 2011 |