Transforming terpene feedstock into polyketide architecture†‡
Philipp
Winter
,
Carine
Vaxelaire
,
Christoph
Heinz
and
Mathias
Christmann
*
TU Dortmund University, Otto-Hahn-Str. 6, D-44227 Dortmund, Germany. E-mail: mathias.christmann@udo.edu; Fax: +49 231 755 5363; Tel: +49 231 755 7854
First published on 10th September 2010
Abstract
The Cu-catalyzed synthesis of skipped 1,4-dienes from allylic acetates and vinyl-Grignard reagents is key to bidirectional modifications of acyclic terpene acetates. As a result, trisubstituted double bond containing subunits can be readily transferred into complex polyketides from inexpensive bulk terpenes.
The focus on target-oriented synthesis of biologically active molecules is shifting toward more economic1 and sustainable chemical processes.2 Among many approaches3 to quantify synthetic efficiency, it has been recently suggested4 along the lines of Hendrickson's (1975) definition5 to use the quotient (construction reactions + strategic redox reactions)/total number of reaction steps as a yardstick for the “ideality” of a given synthesis. We feel that this definition undervalues the importance of minimizing construction steps, i.e. the overall number of C–C-bond forming reactions. Our group's research is dedicated to making larger substructures within natural products6 available from terpene feedstock,7fermentation or catalytic build-up reactions,8e.g. telomerizations.9
As a step toward this goal, we have embarked on the use of oxidized monoterpenes in conjunction with the development of efficient C–C bond forming reactions. Following this loose strategic blueprint, we recently completed the synthesis of englerin A10 from the epoxide of the readily available monoterpene nepetalactone (Fig. 1).
 | ||
| Fig. 1 Terpene epoxides and substructure analysis within ripostatin B. | ||
In this communication, we would like to disclose a strategy for the conversion of epoxygeranyl acetate (1a) and related molecules into 1,4-dienes and representative subsequent conversions into polyketide building blocks using organocatalytic and transition metal catalyzed reactions.
In contrast to terpene biosynthesis which has a sudden increase in complexity through cationic cyclization events, polyketide complexity grows in a rather sequential fashion by two- and three-carbon building blocks. Instead of mimicking the intricate biosynthetic machinery of polyketide synthases, a potential shortcut lies in the possibility to disconnect polyketides into larger fragments. When pondering over the retrosynthesis of ripostatin B,11 a potent RNA polymerase inhibitor,12 we realized that 22 out of its 30 carbon atoms could—in principle—be derived from two molecules of epoxygeranyl acetate (blue), phenyl- and vinylmagnesium bromide (red) as starting materials. The key transformation in such an approach would be the coupling of the Grignard reagents with the allylic acetates.
While the substitution of allylic acetates with phenyl- and alkylmagnesium halides using Li2CuCl4 was precedented by Gansäuer et al.,13 surprisingly, apart from particular examples of Pd-catalyzed couplings with vinylstannanes,14 no general and scalable method for the synthesis of 1,4-dienes15,16 (skipped dienes) from allylic acetates has been reported to our knowledge.
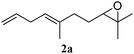
The use of those previously established catalytic conditions (Li2CuCl4 in THF) with vinylmagnesium bromide as the nucleophile met limited success as only 23% of the desired substitution product 2a was detected. After extensive optimization,17 we found that using copper(I)-bromide dimethylsulfide complex (20 mol% in THF) afforded 70% of the desired substitution product. The yield could be further improved by using 1.5 equivalents of vinylmagnesium bromide with copper(I)-iodide (20 mol%) as catalyst in a solvent mixture of tetrahydrofuran/dimethylsulfide (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1) giving rise to 88% of the skipped 1,4-diene 2a (Scheme 1).
∶1) giving rise to 88% of the skipped 1,4-diene 2a (Scheme 1).
 | ||
| Scheme 1 Optimized conditions for the synthesis of 1,4-dienes. | ||
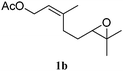
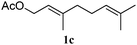
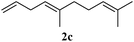
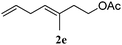
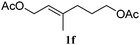
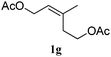
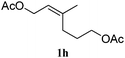
In order to probe the generality (functional group tolerance, integrity of the trisubstituted double bond configuration) of this reaction, we applied our best conditions to the vinyl substitution of a variety of allylic acetates (Table 1). Both epoxygeranyl and epoxyneryl acetate afforded the expected 1,4-dienes with complete retention of the configuration of the allylic double bond (entries 1 and 2) in good to excellent yields. In this instance, the epoxide serves as a handle in subsequent transformations, readily converted to an aldehyde moiety upon treatment with sodium periodate. In addition, their parent derivatives (entries 3 and 4) reacted smoothly to give the desired trienes. In the presence of non-allylic acetates, only those in the allylic position are substituted in good to moderate yield while the others remain unaffected (entries 5 to 8). Furthermore, the presence of free alcohols is well tolerated at the expense of one additional equivalent of Grignard reagent (entries 9 and 10).











In order to showcase the bidirectional modification of epoxydiene 2a and its potential use in the synthesis of polyketides, we converted it into the C5–C15 fragment of ripostatin B (Scheme 2). The four-step synthetic sequence commenced with the cleavage of the epoxide moiety to yield dienal 3. MacMillanet al.18 recently reported the elegant conversion of aldehydes into epoxides using SOMO organocatalysis. Applying this methodology, we were able to isolate the terminal epoxide 5 in an unoptimized 56% yield and with 95% ee. Epoxide opening19 (5 → 6) followed by the oxidative removal of the dithiane20 afforded hydroxyketone 7 in good yield.
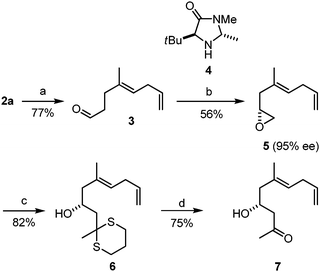 | ||
| Scheme 2
Reagents and conditions: (a) NaIO4, THF/H2O (2∶1), 20 °C; (b) (1) 4·TFA, K2S2O8, LiCl, Cu(TFA)2·xH2O, acetonitrile, water, 4 °C, (2) NaBH4, 0 °C, (3) NaOH, EtOH, H2O, 20 °C; (c) 2-methyl-1,3-dithiane, n-BuLi, THF, 20 °C; (d) PhI(TFA)2, CaCO3, MeOH/acetonitrile (9∶4), 20 °C. | ||
Interestingly, the ripostatin C12–24 subunit 9 bearing a skipped arene-ene moiety can be prepared in a similar approach from epoxygeranyl acetate 1a (Scheme 3). Substitution with phenylmagnesium chloride is followed by periodate cleavage of the epoxide. In order to show the general potential of combining the allylic substitution with an organocatalytic aldol reaction with acetone,21 we obtained the quite advanced fragment 9 in just three synthetic steps from 1a. The major byproduct 10 can be converted to 9 with significantly higher enantioselectivity than for the direct aldol reaction using an oxa-Michael addition developed by List et al.22
 | ||
| Scheme 3
Reagents and conditions: (a) Li2CuCl4, PhMgCl, THF, 0 °C; (b) NaIO4, THF/H2O (2∶1), 20 °C; (c) D-proline, i-PrOH, acetone, 20 °C; (d) 11, Cl3CCO2H, H2O2, dioxane, 32 °C, then P(OEt)3. | ||
Finally, we want to provide an example for the further functionalization of the terminal alkene moiety in 2a. Apart from possible hydroborations, oxidative cleavage and cross-metathesis reactions, we want to use the hydroformylation reaction for the introduction of a free aldehyde functionality on one end of the aliphatic chain while the epoxide on the other terminus remains a masked aldehyde (Scheme 4). Using 2 mol% of Rh(acac)(CO)2 and Xantphos23 as the ligand, we obtained the unbranched aldehyde 12 in virtually quantitative yield. We think that this model compound represents a useful and orthogonally functionalized fragment for the natural product dolabelide D.24

In conclusion, we have developed a simple protocol for the Cu-catalyzed substitution of allylic acetates with vinyl Grignard reagents that is tolerant towards a variety of functional groups. A possible ramification of this method is the reduction of construction steps in the total synthesis of polyketides. In particular, the efficient conversion of terpene acetate derivatives into polyketide building blocks is a harbinger of their increasing importance as cheap and renewable resource in complex molecule synthesis.
Notes and references
- T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010 RSC and references therein.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC.
- (a) S. H. Bertz, J. Am. Chem. Soc., 1982, 104, 5801 CrossRef CAS; (b) I. T. Horvath and P. T. Anastas, Chem. Rev., 2007, 107, 2167 CrossRef CAS; (c) P. L. Fuchs, Tetrahedron, 2001, 57, 6855 CrossRef CAS; (d) F. Qiu, Can. J. Chem., 2008, 86, 903 CrossRef CAS.
- T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657 CrossRef CAS.
- J. B. Hendrickson, J. Am. Chem. Soc., 1975, 97, 5784 CrossRef CAS.
- For an example of this approach, see: C. F. Bender, F. K. Yoshimoto, C. L. Paradise and J. K. De Brabander, J. Am. Chem. Soc., 2009, 131, 11350 Search PubMed.
- K. A. D. Swift, Top. Catal., 2004, 27, 143 CrossRef CAS.
- F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510 CrossRef CAS.
- A. Behr, M. Becker, T. Beckmann, L. Johnen, J. Leschinski and S. Reyer, Angew. Chem., Int. Ed., 2009, 48, 3598 CrossRef CAS.
- M. Willot, L. Radtke, D. Könning, R. Fröhlich, V. H. Gessner, C. Strohmann and M. Christmann, Angew. Chem., Int. Ed., 2009, 48, 9105 CrossRef CAS.
- Isolation: (a) H. Irschik, H. Augustiniak, K. Gerth, G. Höfle and H. Reichenbach, J. Antibiot., 1995, 48, 787 CAS; (b) H. Augustiniak, H. Irschik, H. Reichenbach and G. Höfle, Liebigs Ann., 1996, 1657 Search PubMed; for a synthetic study, see: (c) C. Kujat, M. Bock and A. Kirschning, Synlett, 2006, 449.
- D. Haebich and F. von Nussbaum, Angew. Chem., Int. Ed., 2009, 48, 3397 CrossRef CAS.
- A. Gansäuer, J. Justicia, A. Rosales and B. Rinker, Synlett, 2005, 1954 CrossRef.
- (a) J. Justicia, J. E. Oltra and J. M. Cuerva, J. Org. Chem., 2004, 69, 5803 CrossRef CAS; (b) L. Del Valle, J. K. Stille and L. S. Hegedus, J. Org. Chem., 1990, 55, 3019 CrossRef CAS; (c) A. M. Echavarren and A. M. Castano, Tetrahedron Lett., 1996, 27, 6587; coupling with vinyltrifluoroborates: (d) G. W. Kabalka and M. Al-Masum, Org. Lett., 2006, 8, 11 CrossRef CAS.
- For an excellent overview, see: T. K. Macklin and G. C. Micalizio, Nat. Chem., 2010, 2, 638 Search PubMed and references therein.
- (a) G. W. Kabalka and M. Al-Masum, Org. Lett., 2006, 8, 11 CrossRef CAS; (b) A. Fürstner, O. Larionov and S. Flügge, Angew. Chem., Int. Ed., 2007, 46, 5545 CrossRef; (c) P. S. Diez and G. C. Micalizio, J. Am. Chem. Soc., 2010, 132, 9576 CrossRef CAS.
- See the ESI‡.
- M. Amatore, T. D. Beeson, S. P. Brown and D. W. C. MacMillan, Angew. Chem., 2009, 121, 5223 CrossRef.
- C. W. Wullschleger, J. Gertsch and K.-H. Altmann, Org. Lett., 2010, 12, 1120 CrossRef CAS.
- G. Stork and K. Zhao, Tetrahedron Lett., 1989, 30, 287 CrossRef CAS.
- B. List, P. Pojarliev and C. Castello, Org. Lett., 2001, 3, 573 CrossRef CAS.
- (a) C. M. Reisinger, X. Wang and B. List, Angew. Chem., Int. Ed., 2008, 47, 8112 CrossRef CAS; (b) B. Vakulya, S. Varga, A. Csámpai and T. Soós, Org. Lett., 2005, 7, 1967 CrossRef CAS.
- A. M. Schmidt and P. Eilbracht, J. Org. Chem., 2005, 70, 5528 CrossRef CAS.
- (a) L. Grimaud, R. de Mesmay and J. Prunet, Org. Lett., 2002, 4, 419 CrossRef CAS; (b) N. Desroy, R. Le Roux, P. Phansavath, L. Chiummiento, C. Bonini and J.-P. Genêt, Tetrahedron Lett., 2003, 44, 1763 CrossRef CAS; (c) R. Le Roux, N. Desroy, P. Phansavath and J.-P. Genêt, Synlett, 2005, 429 CAS; (d) G. E. Keck and M. D. McLaws, Tetrahedron Lett., 2005, 46, 4911 CrossRef CAS; (e) P. K. Park, S. J. O'Malley, D. R. Schmidt and J. L. Leighton, J. Am. Chem. Soc., 2006, 128, 2796 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures, optimization of reaction conditions and characterization of compounds. See DOI: 10.1039/c0cc02332a |
| This journal is © The Royal Society of Chemistry 2011 |