Organocatalytic diimide reduction of enamides in water†
Barrie J.
Marsh
,
Emma L.
Heath
and
David R.
Carbery
*
Department of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: d.carbery@bath.ac.uk; Fax: +44 (0)1225 386231; Tel: +44 (0)1225 386144
First published on 16th August 2010
Abstract
Bridged flavinium organocatalysts have displayed efficacy in the diimide mediated reduction of enamides in aqueous conditions. This represents the first diimide reduction of an electron rich alkene and offers a clean alternative to the use of alkylating agents for N-alkylation.
The ability to conduct organic transformations in aqueous media can be viewed to offer a number of advantages. Water is naturally inexpensive, readily available and non-toxic.1,2 In addition, the central role that water plays in the chemistry of life3 offers a fascination to synthetic chemists even though the limited solubility of most organic compounds is deemed synthetically detrimental.
The hydrogenation of alkenes is a reaction of considerable significance in organic synthesis with the majority of alkene reductions being transition metal-catalysed, under heterogeneous or homogeneous conditions.4 However, synthetic problems do exist with traditional heterogeneous hydrogenation methods. Chemo- and regioselectivity in the presence of benzylic functionality, alkene migration and the racemisation of stereocenters have all been reported during the course of attempted hydrogenation reactions.5
Diimide (cis-diazene, HN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH) is a versatile alkene reductant6,7 and, importantly, does not suffer from the synthetic problems discussed above. The majority of synthetic protocols used to prepare diimide involve the in situoxidation of hydrazine.6 An important development in diimide chemistry has been the report of flavinium8 and neutral flavin9 organocatalysts mediating the oxidation of hydrazine to form diimide with subsequent hydrogen transfer from diimide, reducing the alkene and generating nitrogen gas (Scheme 1) which augments the mature oxidative chemistry of flavin catalysts.10
NH) is a versatile alkene reductant6,7 and, importantly, does not suffer from the synthetic problems discussed above. The majority of synthetic protocols used to prepare diimide involve the in situoxidation of hydrazine.6 An important development in diimide chemistry has been the report of flavinium8 and neutral flavin9 organocatalysts mediating the oxidation of hydrazine to form diimide with subsequent hydrogen transfer from diimide, reducing the alkene and generating nitrogen gas (Scheme 1) which augments the mature oxidative chemistry of flavin catalysts.10

Literature precedent strongly suggests that diimide reductions will only proceed on non-polarized alkenes. This assertion is beginning to be challenged with reports of acrylates acting as viable substrates.11 In contrast, there has yet to be a report of a successful diimide reduction of an electron rich alkene such as an enamide. As enamides are accessible through a Brønsted acid catalysed dehydrative condensation of an aldehyde and amide, then the diimide reduction of these alkenes would act as a clean alternative to N-alkylation or metal catalyzed enamide hydrogenation providing a clean alternative to these protocols.
Our laboratory has recently been concerned with the development of new methods for the generation of diimide,12 applications of flavinium catalysts13 and the Ireland–Claisen [3,3]-sigmatropic rearrangements of enamides.14,15 In light of the research synergy presented, we sought to examine the feasibility of a diimide reduction of enamides promoted by bridged-flavinium catalysts (Fig. 1).16
 | ||
| Fig. 1 Flavinium catalysts examined. | ||
It has been demonstrated that flavinium systems 1a–c display improved stability16 with regard to nucleophilic catalyst degradation compared to standard tricyclic flavins, a property expected to be important when trying to reduce difficult electron rich alkene substrates.
The process of optimisation was undertaken with enamide 2a and is presented in Table 1, with solvent, temperature, catalyst, hydrazine loading and reaction concentration all examined.

| Entry | Catalyst | Solvent | NH2NH2·H2O (equiv.) | Conc./M | Conv.a (%) |
|---|---|---|---|---|---|
| a Conversion assayed through 1H NMR analysis of crude reaction mixture. b Reaction conducted at 25 °C. c Reaction conducted at 75 °C. | |||||
| 1b | 1a | CH3CN | 4 | 0.2 | 0 |
| 2b | 1a | DMF | 4 | 0.2 | 0 |
| 3b | 1a | DMSO | 4 | 0.2 | 0 |
| 4c | 1a | CH3CN | 4 | 0.2 | 30 |
| 5c | 1a | DMF | 4 | 0.2 | 10 |
| 6c | 1a | DMSO | 4 | 0.2 | 12 |
| 7c | 1a | H2O | 4 | 0.2 | 42 |
| 8 | 1a | H2O | 4 | 0.2 | 51 |
| 9 | 1b | H2O | 4 | 0.2 | 60 |
| 10 | 1c | H2O | 4 | 0.2 | 31 |
| 11 | 1b | H2O | 8 | 0.2 | 80 |
| 12 | 1b | H2O | 8 | 0.4 | 90 |
| 13 | 1b | H2O | 8 | 0.8 | 84 |
| 14 | 1b | H2O | 8 | 0.5 | 96 |
| 15 | 1b | H2O | 10 | 0.5 | 99 |
| 16 | — | H2O | 10 | 0.5 | 0 |
Initially, solvents examined by Imada were screened, with no reduction observed at room temperature (entries 1–3), however, enamide hydrogenation did begin to occur at elevated temperatures (entries 4–7). Further improvement in reaction efficacy was observed when this reduction reaction was attempted in water at reflux (entry 8). The structure of the catalyst has significant effect on reactivity (entries 8–10) with the 7-CF3 substituted flavinium catalyst 1b offering the highest levels of reactivity (entry 9). By increasing the loading of hydrazine hydrate, the reduction conversion improves further (entry 11). Finally, the reduction was in turn found to be sensitive to concentration with substrate loading at 0.5 M optimal for this reduction (entry 15). Importantly, no reduction was observed in the absence of a flavinium catalyst.
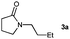
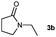
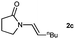
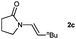
Having optimised the diimide reduction of enamide 2a, a range of enamides were chosen to determine the scope of this reaction under standard conditions (Table 2).
| Entry | Enamide | Product | Conv.b,c (%) |
|---|---|---|---|
a Reaction conditions: 1b (5 mol%), NH2NH2·H2O (10 equiv.), 0.5 M, 100 °C, 18 h.
b Assayed by 1H NMR analysis of crude reaction mixture.
c Isolated yield in parentheses.
d Reaction conducted in H2O/DMSO (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1). ∶1).
|
|||
| 1 |

|
|
100 (93) |
| 2 |

|
|
100 (88) |
| 3 |
|

|
76 |
| 4 |
|

|
100d (91) |
| 5 |
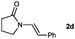
|
|
28c |
| 6 |

|

|
5 |
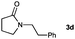
| 7 |

|
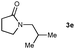
|
31 |
| 8 |
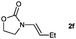
|

|
100 (95) |
| 9 |

|

|
70 |
| 10 |
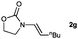
|

|
100d (92) |
| 11 |
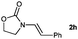
|

|
15 |
| 12 |

|

|
0 |
| 13 |

|
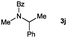
|
16 |
Lactam and oxazolidinone based enamides were observed to reduce efficiently, with 1,2-disubstituted systems producing N-alkyl lactams and oxazolidinones (entries 1–6, 8–10). On achieving full conversion, ready isolation of the N-alkyl products was possible (entries 1, 2, 4, 8 and 10) in excellent yield (see ESI† for full experimental details). Switching the reaction medium to a H2O/DMSO (1∶1) mixture allows for an improved conversion with substrates possessing greater hydrophobic character presumably through improved substrate solubility (entries 4 and 10). The phenyl-substituted substrates 2d (entry 6) and 2h (entry 11) proved challenging. This is consistent with poor solubility and Minnaard's observation that E-stilbene is a poor substrate for diimide alkene reduction.5 Whilst conversion is not optimal, the level of reaction efficiency observed with trisubstituted enamide2e (entry 7) was unexpected. Tri-substituted alkenes are normally poor substrates in diimide reductions of non-polarised alkenes and when examined in combination with the currently discussed enamides, minimal reduction might be expected in this instance. We have observed that 1,1′-disubstituted substrates (entries 12 and 13) are difficult to reduce as gauged using this standard protocol.
It is generally accepted that cis-diimide is highly reactive and readily disproportionates to hydrazine and nitrogen gas.6b In their proposed catalytic cycle, Imada and Naota suggest the catalytic intermediacy of two distinct diimide–flavin complexes with diimide complexed to both oxidised and reduced flavins based upon the stoichiometry of oxygen consumed in these reactions. However, no discussion of how diimide interacts with the flavin catalyst was presented.
The diimide reduction of enamides occur at high temperatures in a strongly H-bonding solvent. We therefore suggest a reversal in order of H-transfer to the enamide in Scheme 2. Slow enamide reduction (5) from the flavin–hydrazine adduct4 is followed by rapid flavin reduction (7) initiated by N2 extrusion from flavin–diazene adduct6.
 | ||
| Scheme 2 Proposed mechanism for hydrogen transfer. | ||
This scheme represents the equivalent of the Imada catalytic anaerobic half-cycle. The overall catalytic cycle can be completed through a similar aerobic half-cycle oxidation of the reduced flavin7. The Imada catalytic cycle involves a second diimide alkene reduction during the aerobic half-cycle which may be productive in this enamide reduction context. However, this enamide reduction requires ca. 8 equivalents of NH2NH2·H2O and may in fact be indicative of non-productive diimide disproportionation under these conditions, ultimately regenerating the flavinium catalyst 1a.
In conclusion, a diimide reduction of electron rich alkenes has been reported for the first time. This synthetic advance has been accomplished in an organocatalytic manner, under aqueous conditions, by using bridged-flavinium catalysts to form diimide from hydrazine and reduce enamides. The presented protocol offers a clean, organocatalytic alternative to N-alkylations, obviating the requirement of alkylating agents or transition metal catalysts. Further studies to explore substrate scope and asymmetric control are ongoing in our laboratories.
We thank the Leverhulme Trust for a project grant (BJM).
Notes and references
- For an excellent review of stereoselective organic reactions in water, see: U. M. Lindström, Chem. Rev., 2002, 102, 2751 Search PubMed.
- For a review of organic reactions on water, see: A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 Search PubMed.
- P. Ball, H2O: A Biography of Water, Phoenix Press, London, 2000 Search PubMed.
- Handbook of Homogeneous Hydrogenation, J. G. de Vries and, C. J. Elsevier, ed., Wiley-VCH, New York, 2007 Search PubMed.
- For an excellent discussion of the potential chemoselectivity and alkene isomerization problems associated with hydrogenation methods and leading references, see: C. Smit, M. W. Fraaije and A. J. Minnaard, J. Org. Chem., 2008, 73, 9482 Search PubMed.
- For reviews concerning the use of diimide, see: (a) S. Hünig, H. R. Müller and W. Thier, Angew. Chem., Int. Ed. Engl., 1965, 4, 271 CrossRef; (b) D. J. Pasto and R. T. Taylor, Org. React., 1991, 40, 91 CAS.
- For recent examples of diimide mediated alkene reduction, see: (a) S. E. Denmark and N. S. Werner, J. Am. Chem. Soc., 2010, 132, 3612 CrossRef CAS; (b) C. D. Smith, J. I. Gavrilyuk, A. J. Lough and R. A. Batey, J. Org. Chem., 2010, 75, 702 CrossRef CAS; (c) C. W. Wullschleger, J. Gertsch and K. Altmann, Org. Lett., 2010, 12, 1120 CrossRef CAS; (d) L. P. Samankumara, M. Zeller, J. A. Krause and C. Brückner, Org. Biomol. Chem, 2010, 8, 1951 RSC; (e) J. F. Teichert and B. L. Feringa, Synlett, 2010, 1200 CAS; (f) C. Singh, A. S. Singh, N. K. Naikade, V. P. Verma, M. Hassam, N. Gupta and S. Pandey, Synlett, 2010, 1014 CAS; (g) J. D. White and J. Yang, Synlett, 2009, 1713 CrossRef CAS; (h) D. P. Dickson, C. Toh, M. Lunda, M. V. Yermolina, D. J. Wardrop and C. L. Landrie, J. Org. Chem., 2009, 74, 9535 CrossRef CAS; (i) M. E. Jung and G. J. Im, J. Org. Chem., 2009, 74, 8739 CrossRef CAS; (j) T. J. Donohoe, R. M. Harris, O. Williams, G. C. Hargaden, J. Burrows and J. Parker, J. Am. Chem. Soc., 2009, 131, 12854 CrossRef CAS; (k) J. R. Harris, S. R. Waetzig and K. A. Woerpel, Org. Lett., 2009, 11, 3290 CrossRef CAS; (l) H. Fuwa, K. Ishigai, T. Goto, A. Suzuki and M. Sasaki, J. Org. Chem., 2009, 74, 4024 CrossRef CAS; (m) T. Yoshikawa, S. Mori and M. Shindo, J. Am. Chem. Soc., 2009, 131, 2092 CrossRef CAS; (n) C. L. Morris, Y. Hu, G. D. Head, L. J. Brown, W. G. Whittingham and R. C. D. Brown, J. Org. Chem., 2009, 74, 981 CrossRef CAS; (o) I. Ortín, J. F. González, E. de la Cuesta and C. Avendaño, Tetrahedron, 2009, 65, 2201 CrossRef CAS; (p) A. Auvinet, B. Eignerová, A. Guy, M. Kotora and T. Durand, Tetrahedron Lett., 2009, 50, 1498 CrossRef CAS; (q) A. Dondoni and A. Marra, Tetrahedron Lett., 2009, 50, 3593 CrossRef CAS; (r) Y. Murakami, M. Yoshida and K. Shishido, Tetrahedron Lett., 2009, 50, 1279 CrossRef CAS.
- Y. Imada, H. Iida and T. Naota, J. Am. Chem. Soc., 2005, 127, 14544 CrossRef CAS.
- Y. Imada, T. Kitagawa, T. Ohno, H. Iida and T. Naota, Org. Lett., 2010, 12, 32 CrossRef.
- For reviews of flavin catalysed oxidations in synthesis, see: (a) F. G. Gelalcha, Chem. Rev., 2007, 107, 3338 CrossRef CAS; (b) Y. Imada and T. Naota, Chem. Rec., 2007, 7, 354 CrossRef CAS.
- P. Lacombe, B. Castagner, Y. Gareau and R. Ruel, Tetrahedron Lett., 1998, 39, 6785 CrossRef CAS.
- B. J. Marsh and D. R. Carbery, J. Org. Chem., 2009, 74, 3186 CrossRef CAS.
- B. J. Marsh and D. R. Carbery, Tetrahedron Lett., 2010, 51, 2136 CrossRef.
- P. M. Ylioja, A. D. Mosley, C. E. Charlot and D. R. Carbery, Tetrahedron Lett., 2008, 49, 1111 CrossRef CAS.
- For a review of enamide substrates in organic synthesis, see: D. R. Carbery, Org. Biomol. Chem., 2008, 6, 3455 Search PubMed.
- (a) W. Li, N. Zhang and L. M. Sayre, Tetrahedron, 2001, 57, 4507 CrossRef CAS; (b) W. Li and L. M. Sayre, Tetrahedron, 2001, 57, 4523 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Representative reaction procedures, experimental data and NMR spectra of novel compounds. See DOI: 10.1039/c0cc02272a |
| This journal is © The Royal Society of Chemistry 2011 |