Revisiting the Kinnel–Scheuer hypothesis for the biosynthesis of palau'amine†‡
Zhiqiang
Ma
,
Jianming
Lu
,
Xiao
Wang
and
Chuo
Chen
*
Department of Biochemistry, UT Southwestern Medical Center at Dallas, 5323 Harry Hines Boulevard, Dallas, TX 75390-9038, USA. E-mail: Chuo.Chen@UTSouthwestern.edu; Fax: 214 648 0320; Tel: 214 648 5048
First published on 16th September 2010
Abstract
We propose herein an alternative biosynthetic pathway for palau'amine in order to resolve the stereochemical issue from the original Kinnel–Scheuer hypothesis. Furthermore, we use this revised hypothesis as a guide toward the laboratory synthesis of palau'amine.
Dimeric pyrrole-imidazole alkaloids1 form a class of structurally unique marine natural products that have, over the past decades, inspired chemists to develop numerous synthetic strategies and methods.2,3 In particular, the recent completion of the syntheses of the [3+2] dimers axinellamines,4 massadines,5 and palau'amine6 by the Baran group stands as a landmark in modern synthesis. The biosynthetic pathway of these pyrrole-imidazole dimers has also been proposed.7 We present herein a plausible biosynthetic pathway of palau'amine (1), and use it as a strategy for the laboratory synthesis of 1.
Kinnel, Scheuer, and co-workers have suggested in their isolation paper that “palau'amine” (2) may arise from a [4+2] cycloaddition of 3-amino-1-(2-aminoimidazolyl)prop-1-ene (3) and 11,12-dehydrophakellin (4), followed by a chloroperoxidase-mediated chlorination and ring contraction (Scheme 1).7c§ However, the structure of palau'amine was recently revised from 2 to 1.8 The C-11/C-12anti stereochemistry in 1 cannot easily be explained by this proposed pathway. The thermal Diels–Alder reaction pathway will result in a syn stereochemistry, and the photo Diels–Alder reaction pathway is not likely considering the lack of sufficient UV-light to the sponges.7a Meanwhile, the difficulty in constructing the piperazine moiety of 1 at a late stage6,9 indicates that this transformation is highly endothermic, and requires significant energy input. We therefore suspected that the ring strain of 1 may be introduced stepwise, both biosynthetically and synthetically.
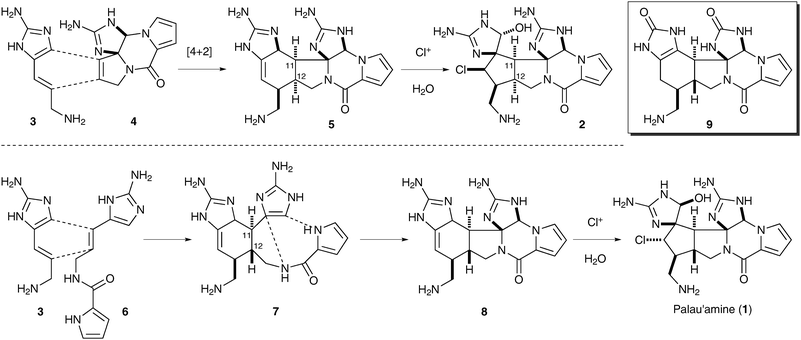 | ||
| Scheme 1 The original (top) and revised (bottom) Kinnel–Scheuer biosynthesis of palau'amine. | ||
In addition to Baran's macrocycle strategy,6 we consider the original Kinnel–Scheuer hypothesis a viable pathway. To reconcile the stereochemical issue of the [4+2] cycloaddition, we propose the intermediacy of ageliferin analog 7 (Scheme 1). Specifically, a [4+2] cycloaddition reaction of 3 and clathrodin (6) would give rise to 7, and set the antiC-11/C-12 stereochemistry. An oxidative bicyclization would then provide the modified Kinnel–Scheuer intermediate 8, which could undergo a chlorinative ring contraction to afford 1. We have synthesized 9, a close analog of 8, in its protected form as a step toward the laboratory synthesis of 1.
Central to our synthetic strategy is a Mn(III)-mediated oxidative radical cyclization reaction,10,11 that, in an intramolecular way, resembles the biogenic [4+2] dimerization to construct the cyclohexenyl core of 7 (Scheme 2). Specifically, oxidation of β-ketoester 15 with Mn(OAc)3 initiated a cascade radical cyclization sequence to deliver 16 that bears the ageliferin skeleton. Two C–C bonds (C-11/C-20 and C-12/C-18) and three stereogenic centers (C-11, C-12, and C-18) were established in this single transformation.
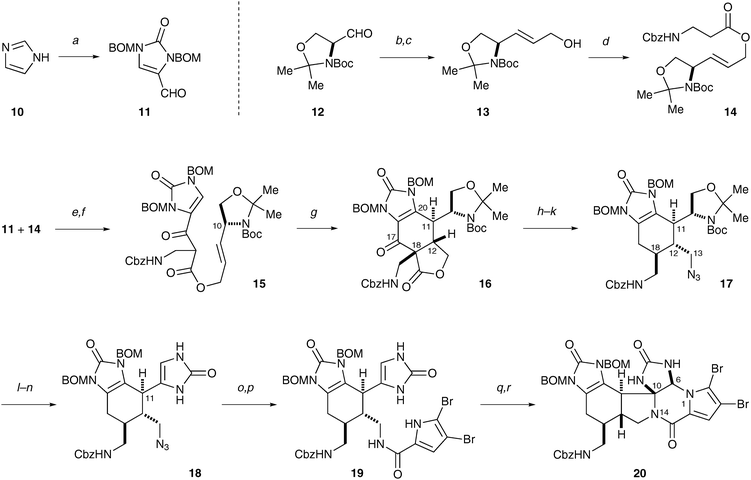 | ||
Scheme 2 The synthesis of 20, a close analog of the Kinnel–Scheuer intermediate. (a) NaH, BOMCl, DMF, 23 °C, then NaH, CuCl2, O2, 23 °C, then POCl3, 23 °C; 68%. (b) Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOOMe, benzene, 23 °C; 95%. (c) DIBAL, THF, 0 °C. (d) Cbz-β-Ala-OH, DCC, CH2Cl2, 23 °C. (e) LiHMDS, THF, −78 °C; 79% over three steps. (f) Dess–Martin periodinane, H2O, CH2Cl2, 23 °C. (g) Mn(OAc)3, HOAc, 60 °C; 36% over two steps. (h) LiOH, THF, H2O, 23 °C. (i) (PhO)2P(O)N3, DEAD, PPh3, THF, 23 °C; 42% over two steps. (j) LiHMDS, THF, then HOAc, −78→0 °C. (k) Ca(BH4)2, THF, 0 °C, then NaBH3CN, HOAc, 50 °C. (l) TFA, CH2Cl2, 0 °C. (m) Dess–Martin periodinane, H2O, CH2Cl2, 23 °C. (n) HCl, MeOH, H2O, 40 °C, then o-(HO2C)C6H4(CO2K), NaOH, KOCN, 110 °C. (o) PPh3, H2O, THF, 80 °C; 7–10% over 6 steps. (p) (Br2-Pyrrole)COCCl3, NEt3, DMF, 70 °C; 33%. (q) PhIO, Na2CO3, TFE, 23 °C. (r) DMSO, 50 °C; 20% over two steps. CHCOOMe, benzene, 23 °C; 95%. (c) DIBAL, THF, 0 °C. (d) Cbz-β-Ala-OH, DCC, CH2Cl2, 23 °C. (e) LiHMDS, THF, −78 °C; 79% over three steps. (f) Dess–Martin periodinane, H2O, CH2Cl2, 23 °C. (g) Mn(OAc)3, HOAc, 60 °C; 36% over two steps. (h) LiOH, THF, H2O, 23 °C. (i) (PhO)2P(O)N3, DEAD, PPh3, THF, 23 °C; 42% over two steps. (j) LiHMDS, THF, then HOAc, −78→0 °C. (k) Ca(BH4)2, THF, 0 °C, then NaBH3CN, HOAc, 50 °C. (l) TFA, CH2Cl2, 0 °C. (m) Dess–Martin periodinane, H2O, CH2Cl2, 23 °C. (n) HCl, MeOH, H2O, 40 °C, then o-(HO2C)C6H4(CO2K), NaOH, KOCN, 110 °C. (o) PPh3, H2O, THF, 80 °C; 7–10% over 6 steps. (p) (Br2-Pyrrole)COCCl3, NEt3, DMF, 70 °C; 33%. (q) PhIO, Na2CO3, TFE, 23 °C. (r) DMSO, 50 °C; 20% over two steps. | ||
To construct 15, we used a convergent approach that comprises an aldol reaction between aldehyde 11 and ester 14 (LiHMDS, THF; 79% over three steps), and an alcohol oxidation (Dess–Martin periodinane, H2O, CH2Cl2). Aldehyde 11 was prepared in one-pot from imidazole (10) (NaH, BOMCl, DMF, then NaH, CuCl2, O2, then POCl3; 68%). Ester 14 was prepared from coupling (DCC, CH2Cl2) of Cbz-β-Ala-OH with the known alcohol 13, which was in turn synthesized in two steps from Garner's aldehyde (12) ((i) Ph3PCHCOOMe, benzene; (ii) DIBAL, THF) according to the literature.12
Treating β-ketoester 15 with Mn(OAc)3 in warm acetic acid results in a quite clean transformation. The ageliferin core skeleton 16 was obtained as the only diastereomer in 36% yield over two steps from the aldol product of 11 and 14.13 The stereochemical course of this reaction was controlled by the C-10 stereogenic center through an A1,3 strain.10 The ester tether, used for the intramolecular Mn(III) oxidation reaction, was then removed. The decarboxylation (LiOH, THF, H2O) of 16 gave rise to a product with cis-C-12/C-18 stereochemistry as the thermodynamic product. The N-14 nitrogen atom was then introduced as an azide by a Mitsunobu reaction ((PhO)2P(O)N3, DEAD, PPh3, THF; 42% for two steps). The C-18 stereocenter was subsequently epimerized through kinetic protonation of the corresponding enolate (LiHMDS, THF, then HOAc) and the C-17 carbonyl group was deoxygenated (Ca(BH4)2, THF,14 then NaBH3CN, HOAc) to afford 17.
In order to implement the oxidative bicyclization strategy to install the piperazine ring, we converted the C-11 side chain of 17 to an imidazolinone group by a three-pot procedure. The acetonide protecting group was first removed (TFA, CH2Cl2) and the resulting alcohol was oxidized (Dess–Martin periodinane, H2O, CH2Cl2). The Boc protecting group was next removed under acidic conditions, followed by in situ treatment of the amino aldehyde with potassium cyanate at pH 4 to afford 18 (HCl, MeOH, H2O, then o-(HO2C)C6H4(CO2K), NaOH, KOCN).15 It is noteworthy that Myers and co-workers have previously reported that amino aldehydes are stable in aqueous media below pH 5, with the aldehyde group existing in its hydrate form.16 In our hands, buffering the aqueous methanol solution of the amino aldehyde with potassium hydrogen phthalate gave the best results.
With 18 in hand, we next sought to introduce the pyrrole group and set the stage for the oxidative bicyclization. The azido group was reduced by a Staudinger reduction (PPh3, H2O, THF) and the product was obtained in 7–10% yield over 6 steps after HPLC purification. The pyrrole group was then installed ((Br2-Pyrrole)COCCl3, NEt3, DMF; 33%). Use of a less pure amine for this reaction resulted in significantly less or no product.
We previously reported a hypervalent iodine(III)-mediated oxidative bicyclization reaction of dihydro-14-oxo-oroidin to give dibromophakellstatin in quantitative yield.17 Pleasingly, use of the same reaction conditions (PhI(OAc)2, Na2CO3, TFE) to oxidize 19 did afford 20, though inconsistently in 0–5% yield. After extensive optimization, we found PhIO to be a better oxidant, which gave rise to an unstable product that slowly converted to the Kinnel–Scheuer intermediate analog 20 at elevated temperature in DMSO in 20% yield over two steps. A similar unstable intermediate, which was suspected to be the monocyclized product, was also observed by Büchi and co-workers when oxidizing dihydrooroidin with bromine to form dibromophakellin.15b,18
In summary, as part of our palau'amine synthesis project, we have synthesized a close analog of the modified Kinnel–Scheuer intermediate from their biosynthesis proposal. The stereochemistry of this advanced synthetic intermediate is consistent with the revised structure of palau'amine. The biomimetic oxidative ring contraction of the Kinnel–Scheuer intermediate has previously been realized in model systems in the laboratory by Romo,3d Lovely,3c Baran19 and us.10 In particular, the method developed by Romo allowed for the introduction of the chlorine atom. We are currently examining the practicality of these methods in converting advanced synthetic intermediates such as 20 into palau'amine (1).
We thank the NIH (Grant NIGMS R01-GM079554), the Welch Foundation (I-1596), and UT Southwestern for financial support, and Prof. Joseph Fox (University of Delaware) for the suggestion of the use of ethyltrichlorosilane-deactivated silica gel.
Notes and references
- B. Forte, B. Malgesini, C. Piutti, F. Quartieri, A. Scolaro and G. Papeo, Mar. Drugs, 2009, 7, 705–753 CrossRef CAS.
- (a) T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657–4673 CrossRef CAS; (b) B. Heasley, Eur. J. Org. Chem., 2009, 1477–1489 CrossRef CAS; (c) H.-D. Arndt and M. Riedrich, Angew. Chem., Int. Ed., 2008, 47, 4785–4788 CrossRef CAS; (d) M. Köck, A. Grube, I. B. Seiple and P. S. Baran, Angew. Chem., Int. Ed., 2007, 46, 6586–6594 CrossRef; (e) D. E. N. Jacquot and T. Lindel, Curr. Org. Chem., 2005, 9, 1551–1565 CrossRef CAS; (f) H. Hoffmann and T. Lindel, Synthesis, 2003, 1753–1783 CAS.
- For example: (a) L. E. Overman, B. N. Rogers, J. E. Tellew and W. C. Trenkle, J. Am. Chem. Soc., 1997, 119, 7159–7160 CrossRef CAS; (b) J. T. Starr, G. Koch and E. M. Carreira, J. Am. Chem. Soc., 2000, 122, 8793–8794 CrossRef CAS; (c) C. J. Lovely, H. Du and H. V. R. Dias, Org. Lett., 2001, 3, 1319–1322 CrossRef CAS; (d) A. S. Dilley and D. Romo, Org. Lett., 2001, 3, 1535–1538 CrossRef CAS; (e) I. Kawasaki, N. Sakaguchi, N. Fukushima, N. Fujioka, F. Nikaido, M. Yamashita and S. Ohta, Tetrahedron Lett., 2002, 43, 4377–4380 CrossRef CAS; (f) S. G. Koenig, S. M. Miller, K. A. Leonard, R. S. Löwe, B. C. Chen and D. J. Austin, Org. Lett., 2003, 5, 2203–2206 CrossRef CAS; (g) P. S. Baran, A. L. Zografos and D. P. O'Malley, J. Am. Chem. Soc., 2004, 126, 3726–3727 CrossRef; (h) P. S. Baran, D. P. O'Malley and A. L. Zografos, Angew. Chem., Int. Ed., 2004, 43, 2674–2677 CrossRef CAS; (i) V. B. Birman and X.-T. Jiang, Org. Lett., 2004, 6, 2369–2371 CrossRef CAS; (j) H. Garrido-Hernandez, M. Nakadai, M. Vimolratana, Q. Li, T. Doundoulakis and P. G. Harran, Angew. Chem., Int. Ed., 2005, 44, 765–769 CrossRef CAS; (k) I. Kawasaki, N. Sakaguchi, A. Khadeer, M. Yamashita and S. Ohta, Tetrahedron, 2006, 62, 10182–10192 CrossRef CAS; (l) M. S. Bultman, J. Ma and D. Y. Gin, Angew. Chem., Int. Ed., 2008, 47, 6821–6824 CrossRef CAS; (m) J. Hudon, T. A. Cernak, J. A. Ashenhurst and J. L. Gleason, Angew. Chem., Int. Ed., 2008, 47, 8885–8888 CrossRef CAS.
- (a) J. Yamaguchi, I. B. Seiple, I. S. Young, D. P. O'Malley, M. Maue and P. S. Baran, Angew. Chem., Int. Ed., 2008, 47, 3578–3580 CrossRef CAS; (b) D. P. O'Malley, J. Yamaguchi, I. S. Young, I. B. Seiple and P. S. Baran, Angew. Chem., Int. Ed., 2008, 47, 3581–3583 CrossRef CAS.
- S. Su, I. B. Seiple, I. S. Young and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 16490–16491 CrossRef CAS.
- I. B. Seiple, S. Su, I. S. Young, C. A. Lewis, J. Yamaguchi and P. S. Baran, Angew. Chem., Int. Ed., 2009, 49, 1095–1098 CrossRef.
- (a) R. P. Walker, D. J. Faulkner, D. V. Engen and J. Clardy, J. Am. Chem. Soc., 1981, 103, 6772–6773 CrossRef CAS; (b) P. A. Keifer, R. E. Schwartz, M. E. S. Koker, J. R. G. Hughes, D. Rittschof and K. L. Rinehart, J. Org. Chem., 1991, 56, 2965–2975 CrossRef CAS; (c) R. B. Kinnel, H.-P. Gehrken, R. Swali, G. Skoropowski and P. J. Scheuer, J. Org. Chem., 1998, 63, 3281–3286 CrossRef CAS; (d) A. Al Mourabit and P. Potier, Eur. J. Org. Chem., 2001, 237–243 CrossRef CAS; (e) Ref. 3h ; (f) B. H. Northrop, D. P. O'Malley, A. L. Zografos, P. S. Baran and K. N. Houk, Angew. Chem., Int. Ed., 2006, 45, 4126–4130 CrossRef CAS; (g) C. Pöverlein, G. Breckle and T. Lindel, Org. Lett., 2006, 8, 819–821 CrossRef.
- (a) M. S. Buchanan, A. R. Carroll, R. Addepalli, V. M. Avery, J. N. A. Hooper and R. J. Quinn, J. Org. Chem., 2007, 72, 2309–2317 CrossRef CAS; (b) M. S. Buchanan, A. R. Carroll and R. J. Quinn, Tetrahedron Lett., 2007, 48, 4573–4574 CrossRef CAS; (c) A. Grube and M. Köck, Angew. Chem., Int. Ed., 2007, 46, 2320–2324 CrossRef CAS; (d) H. Kobayashi, K. Kitamura, K. Nagai, Y. Nakao, N. Fusetani, R. W. M. v. Soest and S. Matsunaga, Tetrahedron Lett., 2007, 48, 2127–2129 CrossRef CAS.
- Z. Ma and C. Chen, unpublished results.
- X. Tan and C. Chen, Angew. Chem., Int. Ed., 2006, 45, 4345–4348 CrossRef CAS.
- (a) B. B. Snider, Chem. Rev., 1996, 96, 339–363 CrossRef CAS; (b) G. G. Melikyan, Org. React., 1997, 49, 427–675 CAS.
- (a) S. Lin, Z.-Q. Yang, B. H. B. Kwok, M. Koldobskiy, C. M. Crews and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 6347–6355 CrossRef CAS; (b) L. Devel, L. Hamon, H. Becker, A. Thellend and A. Vidal-Cros, Carbohydr. Res., 2003, 338, 1591–1601 CrossRef CAS.
- While the crude 1H NMR indicated 16 to be the only significant product, it was not stable to silica gel column chromatography, leading to its diminished isolated yield. The stereochemistry of 16 was assigned based on the stereochemical studies from ref. 10, and confirmed by ROESY experiments of subsequent synthetic intermediates.
- (a) J. Kollonitsch, O. Fuchs and V. Gábor, Nature, 1954, 173, 125–126 CAS; (b) H. C. Brown, S. Narasimhan and Y. M. Choi, J. Org. Chem., 1982, 47, 4702–4708 CrossRef CAS.
- (a) K. J. Wiese, K. Yakushijin and D. A. Horne, Tetrahedron Lett., 2002, 43, 5135–5136 CrossRef CAS. See also: (b) L. H. Foley and G. Büchi, J. Am. Chem. Soc., 1982, 104, 1776–1777 CrossRef CAS; (c) Ref. 3g .
- A. G. Myers, D. W. Kung and B. Zhong, J. Am. Chem. Soc., 2000, 122, 3236–3237 CrossRef CAS.
- (a) J. Lu, X. Tan and C. Chen, J. Am. Chem. Soc., 2007, 129, 7768–7769 CrossRef CAS. See also: (b) K. S. Feldman and A. P. Skoumbourdis, Org. Lett., 2005, 7, 929–931 CrossRef CAS; (c) Ref. 15a ; (d) Ref. 15b .
- Besides Büchi's proposed formation of a C-10/N-14 linkage in ref. 15b for his dibromophakellin synthesis, it is also possible that a N-1/C-6 macrocycle, resembling Baran's palau'amine intermediate 4′ in ref. 6, was formed during the oxidation of 19.
- D. P. O'Malley, K. Li, M. Maue, A. L. Zografos and P. S. Baran, J. Am. Chem. Soc., 2007, 129, 4762–4775 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and characterization data. See DOI: 10.1039/c0cc02214d |
| § This biosynthetic pathway was a “better alternative” suggested by a reviewer of ref. 7c. |
| This journal is © The Royal Society of Chemistry 2011 |