Porous protein frameworks with unsaturated metal centers in sterically encumbered coordination sites†‡
Robert J.
Radford
a,
Morgan
Lawrenz
ab,
Phuong C.
Nguyen
a,
J. Andrew
McCammon
abcd and
F. Akif
Tezcan
*a
aDepartment of Chemistry and Biochemistry, University of California, San Diego 9500 Gilman Dr, La Jolla, CA 92037, USA. E-mail: tezcan@ucsd.edu
bCenter for Theoretical Biological Physics, University of California, San Diego 9500 Gilman Dr, La Jolla, CA 92037, USA
cDepartment of Pharmacology, University of California, San Diego 9500 Gilman Dr, La Jolla, CA 92037, USA
dHoward Hughes Medical Institute, University of California, San Diego 9500 Gilman Dr, La Jolla, CA 92037, USA
First published on 25th August 2010
Abstract
Described is an engineered metal-binding protein, MBPPhen2, which forms porous crystalline frameworks that feature coordinatively unsaturated Zn- and Ni-centers.
Nature predominantly uses amino acids to construct cellular machinery and advanced materials due to the structural and functional diversity these 20 building blocks provide, which has motivated chemists to follow suit. On the peptide level, there have been impressive engineering efforts over the last two decades. These efforts produced, among others, protein-like assemblies containing inorganic functional groups,1 and materials with diverse applications in tissue engineering,2nanoparticle assembly3 and molecular electronics.4 Yet, the mastery over the self-assembly of peptide-based structures has not yet reached a level where discrete, multi-dimensional architectures can be engineered with ease beyond one-dimensional, fibrillar structures, owing to the fact that peptide building blocks are flexible or prone to heterogeneous aggregation.5 These drawbacks could be circumvented if rigid, folded proteins are used as building blocks, and accordingly, there have been impressive, but isolated cases of success in the rational design of discrete protein assemblies.6–8 The structural order and the chemical functionalizability of such multi-protein assemblies as virus capsids, protein cages and layers are now being widely exploited to generate hybrid materials toward catalysis,9 imaging,10 templating/patterning,11 and drug delivery.12 In parallel, nano/meso-porous crystalline protein frameworks are showing promise towards applications such as molecular separation13 and heterogeneous catalysis14 in a similar fashion as zeolites and metal–organic frameworks (MOFs).
Given the potential advantages of proteins as building blocks for functional materials, we recently endeavored to develop bottom-up strategies toward directing protein self-assembly (PSA).15 Inspired by advances in supramolecular coordination chemistry, we reasoned that the strength and reversibility of metal coordination interactions can be exploited to guide PSA under thermodynamic control, while simultaneously enabling the incorporation of inorganic functionalities into multi-protein assemblies and frameworks. Along these lines, we describe here an engineered protein (MBPPhen2) based on the four-helix bundle hemeprotein cytochrome cb562 that forms porous crystalline frameworks upon metal coordination and features unsaturated metal centers toward potential applications in catalysis.
We have previously shown that cyt cb562 variants with a proper arrangement of surface histidines (His) can be assembled through metal coordination into discrete oligomers whose shapes and symmetries are entirely dictated by the stereochemical preference of the added metal ions.15 Analogously to MOFs, however, these metal ions are coordinatively saturated, and while necessary for the structural integrity of the protein assemblies, they are functionally “silent”. To broaden the scope of Metal-Directed Protein Self-Assembly (MDPSA), we have recently incorporated non-natural metal chelates onto the surface of Helix3 of cyt cb562.16 One resulting construct, MBPPhen1 (Fig. 1), with a 1,10′-phenanthroline (Phen) derivative attached to an engineered surface Cys (C59), was found to form Ni-induced trimers, whereby the interfacial Ni centers were coordinated to a single Phen from one protomer and a histidine (H77) from a second, with two open coordination sites filled by solvent molecules (Fig. S3.1, ESI†). The open Ni coordination was a consequence of the Phen moiety being buried in a surface crevice under the overhang formed by the 50's loop, which prevented the formation of the saturated Ni:MBPPhen13 complex. Significantly, the open, trimeric Ni3:MBPPhen13 architectures were found to stack up in the crystal lattice, giving rise to porous 3-D frameworks.
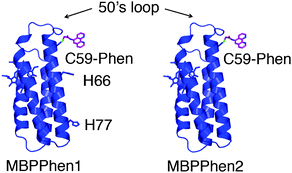 | ||
| Fig. 1 Cartoon representation of MBPPhen1 and MBPPhen2. | ||
In this study, we sought to understand (1) if there is a thermodynamic bias towards the burial of Phen under the 50's loop (i.e., if this surface feature can be reproducibly used as steric bulk in analogy to smaller, sterically encumbered ligand frameworks), and (2) if this burial can yield alternative protein oligomers/frameworks with unsaturated metal centers. To this end, we made a variant of MBPPhen1 (MBPPhen2, Fig. 1) that lacks other likely surface ligands on Helix3 including H77 and H66.
An important starting point was to determine whether the Phen burial would occur in the absence of metal coordination. MBPPhen2 was thus crystallized in the presence of EDTA and its structure determined at 2.1 Å resolution (PDB ID: 3NMI). The asymmetric unit of the C2 symmetric lattice contains six MBPPhen2 monomers arranged in three pairs (Fig. S3.2, ESI†). Two of the pairs are identical to each other and feature a head-to-head alignment of monomers, where the Phen moieties form π-stacking interactions with one another while still buried under the 50's loop (Fig. 2a). Significantly, the conformation of these 4 Phen groups in the asymmetric unit is identical to that observed in the Ni3:MBPPhen13 complex (Fig. 2b): the aromatic moiety packs tightly into the cavity lined by the side chains of T44, P53, M58, F61 and A62, and the amide nitrogen of the linker between C59 and Phen forms a H-bond with the backbone carbonyl of P53. The remaining pair of MBPPhen2 molecules also forms a head-to-head dimer, but in contrast, their Phen groups are now observed outside of the surface crevice. One Phen group is found pointing away from the 50's loop lying flat against the Helix3 surface, whereas the other one is significantly disordered as suggested by a weak corresponding electron density but still forming some π-stacking interactions with the former.
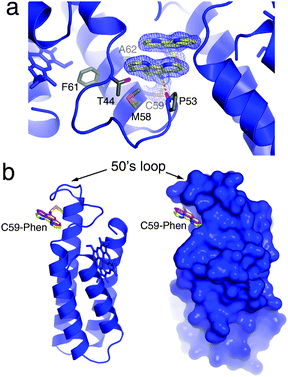 | ||
| Fig. 2 (a) Crystal structure of metal-free MBPPhen2, highlighting the Phen moiety buried under the 50's loop as well as its π-stacking interactions with a Phen group from another MBPPhen2 molecule in the asymmetric unit. The 2Fo − Fc map is contoured at 1.5σ. (b) Ribbon and surface representations of metal-free MBPPhen2, showing that its Phen conformation (yellow) is the same as that observed in the Ni3:MBPPhen13 structure (magenta). | ||
While the crystal structure of metal-free MBPPhen2 indicates a preference of the Phen moiety to be buried under the 50's loop, it also shows that it can explore “out” conformations. To gain further insight into the energetics of Phen–protein surface interactions, we carried out alchemical free energy calculations using the thermodynamic cycle in Fig. S3.3 (ESI†). These calculations indicate that the buried Phen conformation is 4.2 ± 1.3 kcal mol−1 more favorable than an extended and fully solvent-exposed conformation.
We next examined if the steric encumbrance around the Phen group in MBPPhen2 affects its coordination behaviour. Specifically, we investigated the solution oligomerization state of MBPPhen2 in the presence of Ni2+, which would be expected to give a trimeric species for a fully exposed Phen group. Sedimentation velocity (SV) experiments with MBPPhen2 in the presence of a 1/3 equivalent of Ni2+ to drive the formation of the Ni(MBPPhen2)3 instead indicate that the predominant species in solution is a dimer (Fig. 3), even at a protein/Ni concentration of 600/200 μM.
![Sedimentation velocity distributions of MBPPhen2 in the absence of metal and presence of Ni2+ ([MBPPhen2] = 600 μM, [Ni2+] = 200 μM).](/image/article/2011/CC/c0cc02168g/c0cc02168g-f3.gif) | ||
| Fig. 3 Sedimentation velocity distributions of MBPPhen2 in the absence of metal and presence of Ni2+ ([MBPPhen2] = 600 μM, [Ni2+] = 200 μM). | ||
The solution oligomerization behavior of MBPPhen2 is paralleled in the solid state. We obtained crystals of the Ni2+ and Zn2+ adducts of MBPPhen2 and determined their structures at 3.1 and 2.8 Å resolution, respectively (PDB IDs: 3NMJ and 3NMK). Remarkably, the hexagonal (P65 space group) crystals of the Ni2+ and Zn2+ complexes are entirely isostructural despite the distinct stereochemical preferences of the two ions. The Ni2+ and Zn2+-induced MBPPhen2 dimers are shown in Fig. 4a. These C2-symmetric V-shaped dimers are mediated solely by Ni/Zn binding to Phen groups, one of which is in the buried and the other in an extended conformation. Ni and Zn are found in identical distorted trigonal bipyramidal geometries in Λ configuration, which are completed by a water molecule (Fig. 4b). In the asymmetric unit, pairs of Ni- and Zn-induced dimers are further interlaced, yielding D2-symmetric tetramers that hold the metal centers in close proximity (7 Å between metal centers, 3 Å between coordinated water molecules, Fig. 4c and d). Clearly, the combination of the steric bulk around the Phen groupsand lattice packing interactions are ultimately responsible for the formation of this particular supramolecular arrangement, and force Ni2+ and Zn2+ to adopt the same coordination geometry. The enforcement of identical coordination geometries on metal ions with distinct stereochemical preferences is typically reserved for rigid, bulky ligand platforms,17 which include highly evolved protein scaffolds with internal coordination sites. The fact that this can be achieved on the surface of a protein highlights the potential of crystalline protein frameworks being used as platforms for metal-based catalysis, as elegantly demonstrated in small-molecule crystal systems.18 Further in support for such potential applications, the crystals of Ni:MPBPhen22 and Zn:MPBPhen22 are highly porous (Fig. 4e), with two sets of hexagonal channels 6 and 2 nm wide and an overall solvent content of 61%. A single asymmetric unit that contains a dimer of MBPPhen2 dimers is highlighted within this lattice. These tetrameric units are arranged into columns using helix–helix packing interactions and form the lining of the two hexagonal channels (Fig. S3.4, ESI†). A surface representation of these MBPPhen2 columns indicates that the interfacial metal centers should be accessible from the solvent channels, yet protected enough to potentially display selectivity.
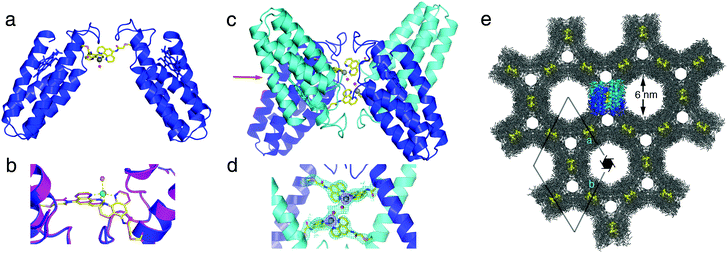 | ||
| Fig. 4 (a) Zn:MPBPhen22 crystal structure. The analogous Ni complex is isostructural with the Zn complex (rmsd = 0.293 Å over all Cα's). (b) Metal coordination environment in Zn (blue) and Ni (magenta) complexes. Zn and Ni centers are shown as green and yellow spheres, respectively. (c) Dimer of Zn:MPBPhen22 dimers in the asymmetric unit. (d) Metal coordination environments in the asymmetric unit of Zn:MPBPhen22 crystals, showing the proximity of the neighboring metal centers. The 2Fo − Fc map is contoured at 1.2σ. (e) Zn:MPBPhen22 crystal lattice viewed down the c-axis. The highlighted dimer of dimers is viewed along the direction indicated with a red arrow in (c). Phen groups are shown as yellow spheres to highlight the relative positions of metal centers in the lattice. | ||
We are currently in the process of investigating whether the same isostructural units can be obtained in the presence of other metals, and testing the reactivity of Ni:MPBPhen22 and Zn:MPBPhen22 single crystals. As a prelude to these studies, we subjected these crystals to chemical crosslinking with 6% (v/v) glutaraldehyde for 30 min. Whereas unmodified crystals immediately dissolve upon transfer from the precipitation solution (30% PEG400, 0.2 M ammonium sulfate) into water, the crosslinked crystals are indefinitely stable in water, even after being kept at 98 °C for 10 min, or in a 50% acetonitrile/water mixture (Fig. S3.5–S3.8, ESI†).
In our efforts to control protein self-assembly through metal coordination, we have benefited from our deconstruction of proteins as simple, rigid ligand platforms for metal coordination. The study undertaken here has revealed strong parallels between the coordination complexes/frameworks of small organic molecules and those of proteins, suggesting that the approach of using proteins as metal ligands may have utilities beyond simply controlling their self-assembly.
R.J.R., P.C.N. and F.A.T. were supported by DOE (DE-FG02-10ER46677, crystallographic analyses), NSF (CHE-0908115, protein design/modification), an NIH traineeship (R.J.R.), and the Beckman Foundation. M.L. and J.A.M. were supported in part by NSF, NIH, HHMI, CTBP and NBCR. Portions of this research were carried out at SSRL, operated by Stanford University on behalf of DOE.
Notes and references
- A. Lombardi, C. M. Summa, S. Geremia, L. Randaccio, V. Pavone and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6298–6305 CrossRef CAS.
- T. C. Holmes, S. de Lacalle, X. Su, G. S. Liu, A. Rich and S. G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6728–6733 CrossRef CAS.
- M. M. Stevens, N. T. Flynn, C. Wang, D. A. Tirrell and R. Langer, Adv. Mater., 2004, 16, 915–918 CrossRef CAS.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS.
- E. H. C. Bromley, K. Channon, E. Moutevelis and D. N. Woolfson, ACS Chem. Biol., 2008, 3, 38–50 CrossRef CAS.
- J. E. Padilla, C. Colovos and T. O. Yeates, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2217–2221 CrossRef CAS.
- P. Ringler and G. E. Schulz, Science, 2003, 302, 106–109 CrossRef CAS.
- S. Burazerovic, J. Gradinaru, J. Pierron and T. R. Ward, Angew. Chem., Int. Ed., 2007, 46, 5510–5514 CrossRef CAS.
- S. Abe, K. Hirata, T. Ueno, K. Morino, N. Shimizu, M. Yamamoto, M. Takata, E. Yashima and Y. Watanabe, J. Am. Chem. Soc., 2009, 131, 6958–6960 CrossRef CAS.
- J. D. Lewis, G. Destito, A. Zijlstra, M. J. Gonzalez, J. P. Quigley, M. Manchester and H. Stuhlmann, Nat. Med. (N. Y.), 2006, 12, 354–360 CrossRef CAS.
- D. Moll, C. Huber, B. Schlegel, D. Pum, U. B. Sleytr and M. Sara, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14646–14651 CrossRef CAS.
- M. L. Flenniken, L. O. Liepold, B. E. Crowley, D. A. Willits, M. J. Young and T. Douglas, Chem. Commun., 2005, 447–449 RSC.
- L. Z. Vilenchik, J. P. Griffith, N. St Clair, M. A. Navia and A. L. Margolin, J. Am. Chem. Soc., 1998, 120, 4290–4294 CrossRef CAS.
- T. Koshiyama, N. Kawaba, T. Hikage, M. Shirai, Y. Miura, C. Y. Huang, K. Tanaka, Y. Watanabe and T. Ueno, Bioconjugate Chem., 2010, 21, 264–269 CrossRef CAS.
- E. N. Salgado, R. J. Radford and F. A. Tezcan, Acc. Chem. Res., 2010, 43, 661–672 CrossRef CAS.
- R. J. Radford and F. A. Tezcan, J. Am. Chem. Soc., 2009, 131, 9136–9137 CrossRef CAS.
- E. E. Benson, A. L. Rheingold and C. P. Kubiak, Inorg. Chem., 2010, 49, 1458–1464 CrossRef CAS.
- Z. Huang, P. S. White and M. Brookhart, Nature, 2010, 465, 598–601 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and additional figures and tables. See DOI: 10.1039/c0cc02168g |
| ‡ This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| This journal is © The Royal Society of Chemistry 2011 |