Synthesis and characterization of an infinite sheet of metal–alkyl bonds: unfolding the elusive structure of an unsolvated alkali-metal trisalkylmagnesiate†‡
Sharon E.
Baillie
a,
William
Clegg
b,
Pablo
García-Álvarez
a,
Eva
Hevia
*a,
Alan R.
Kennedy
a,
Jan
Klett
a and
Luca
Russo
b
aWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, UK G1 1XL. E-mail: eva.hevia@strath.ac.uk
bSchool of Chemistry, Newcastle University, Newcastle upon Tyne, UK NE1 7RU
First published on 31st August 2010
Abstract
The study of co-complexation reactions between NaCH2SiMe3 and Mg(CH2SiMe3)2 has allowed the isolation and structural elucidation of the first solvent-free alkali-metal alkylmagnesiate [{NaMg(CH2SiMe3)3}∞] (1) as well as the related solvent-free sodium alkyl [{(NaCH2SiMe3)4}∞] (3).
Introduced by Wittig almost 60 years ago,1 alkali-metal magnesiates constitute an important family of organometallic reagents which have remained for most of this time in the domain of inorganic structural chemists. However, during the past decade, these bimetallic species have also captured widespread interest amongst the organic synthetic community due to their unique chemical profiles.2 Carriers of an enhanced reactivity and exceptional functional group tolerance, combined with selectivity patterns distinct from those displayed by their monometallic components, these ates play a key role in many important organic transformations. Amongst them, homoleptic alkyl magnesiates (which, depending on their stoichiometry, can be grouped as triorganomagnesiates MMgR3 or tetraorganomagnesiates M2MgR4, where M = alkali metal, R = alkyl group) have proved to be highly versatile reagents, finding extensive application not only in magnesium–halogen exchange and deprotonation reactions but also in alkylation reactions of ketones due to their exceptional carbonucleophilicity.2
Despite some structural studies on solvated alkali-metal magnesiates (including pioneering contributions by Weiss that have advanced the understanding of the special reactivity of these compounds),3 to date the structures of the unsolvated compounds have proved elusive. In monometallic chemistry, unsolvated s-block organometallic compounds generally exist as oligomeric aggregates whose reactivity can be finely tuned by addition of Lewis bases that can modify their degrees of aggregation. In addition, for bimetallic (ate) compounds, their unique (synergic) reactivities have been linked to the presence of bridging ligands that will bring the alkali-metal and magnesium into close proximity to each other, enabling effective metal–ligand–metal communication.4
To fill an important gap in the knowledge of bimetallic chemistry, we provide here unprecedented insight into the constitution and stability of unsolvated alkali-metal magnesiates, by exploring co-complexation reactions of sodium and magnesium alkyls in non-polar solvents. We present the first structural characterization of a donor-free alkali-metal trisalkylmagnesiate [(NaMgR3)∞] (1) (R = CH2SiMe3) and findings of a study of the aggregation of these compounds in C6D6 solutions using 1H diffusion-ordered NMR spectroscopy (1H DOSY) which provides a greater understanding of the real constitution of these compounds in solution.
Firstly we examined the reaction of equimolar amounts of monometallic alkyls NaCH2SiMe3 and Mg(CH2SiMe3)2 using bulk hexane as a solvent, which afforded a white precipitate. Addition of toluene dissolved this solid to give a colourless solution. This enhanced solubility as well as the lack of colour in the solution was indicative of the formation of a mixed-metal compound, since both monometallic components are totally insoluble in this solvent mixture and, even more revealingly, NaCH2SiMe3 readily deprotonates toluene at room temperature to yield NaCH2Ph which precipitates as an orange solid. On cooling, the colourless solution deposited crystals of the sodium magnesiate [{NaMg(CH2SiMe3)3}∞] (1) as determined by NMR spectroscopy and X-ray crystallography. A diagnostic resonance at −1.71 ppm is observed for the trimethylsilylmethyl CH2 group, significantly downfield from that found for NaCH2SiMe3 (−2.44 ppm) suggesting that the alkyl groups in 1 possess a lesser carbanionic character than in the sodium compound.5
The mixed-metal composition of 1 was confirmed by an X-ray crystallographic study (Fig. 1).§ Its basic repeat unit comprises a trigonal planar magnesium bonded to three alkyl groups. One CH2SiMe3 ligand bridges magnesium and sodium in the asymmetric unit (Na–C5 2.6708(19) Å, Mg–C5 2.1617(19) Å, Fig. 1a), whereas the remaining two alkyl ligands bond to sodium atoms of neighbouring units giving rise to an intricate two-dimensional honeycomb sheet structure which contains 12-atom {(NaCMgC)3} fused rings (Fig. 1b) with the sterically hindered SiMe3 groups alternately binding to each face of the sheet. A space filling model of 1 (Fig. 1c) shows that the latter provide a protective steric shelter of the polar (reactive) metal–carbon bonds. Each of these rings accommodates 6 metals (3 Na, 3 Mg) and 6 alkyl ligands and is interconnected with another six rings within the polymeric structure. This intriguing fused ring assembly is probably directed by sodium, which in order to attain a higher coordination number, under solvent-free conditions, needs to interact with three alkyl groups. In terms of supramolecular chemistry 1 can be envisaged as a combination of trigonal nodes (sodium and magnesium centres) connected by alkyl ligands (spacers),6 giving rise to a 2D infinite network in which all alkyl groups are equivalent, acting as magnesium and sodium linkers. The analysis of the different metal–carbon bond distances7 revealed that there is no significant difference that would define a specific molecular unit, indicating that this extended sheet structure is the result of a regular arrangement of Na and Mg cations that are held together by a combination of relatively strong (short) electron-deficient Na–C and Mg–C bonds (whose lengths are similar to those found in discrete ate molecules).8
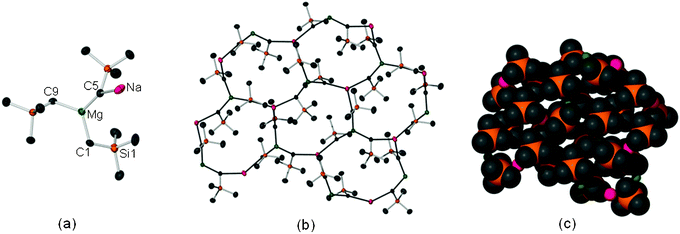 | ||
| Fig. 1 (a) Asymmetric unit of 1. (b) Polymeric sheet network of 1. (c) Space filling model for polymeric structure of 1 (in all the figures hydrogen atoms have been omitted for clarity). | ||
To the best of our knowledge, the unique 2D organometallic honeycomb structure of 1 constitutes the first example of a solvent-free alkali-metal trisalkylmagnesiate to be structurally defined. This ring-fused assembly is rather rare in general s-block chemistry, being previously found for lithiated (organosulfonyl)acetonitriles and cyanophosphonates.9 However, the association between the distinct asymmetric units in these compounds takes place via dative bonds between lithium centres and heteroatomic substituents of the anionic ligands, whereas in 1 the metallic ions are connected by highly polar (and therefore potentially more reactive!) electron deficient Na–C and Mg–C bonds. The infinitely aggregated structure of 1 contrasts with solvated structures of related alkyl magnesiates which, depending on the hapticity and donor ability of the Lewis bases, can exhibit a contacted ion pair structure (usually monomers or dimers with a linear “Weiss” alkali-metal magnesiate motif)3 or a solvent separated ion pair structure, where no alkali-metal–magnesium communication is possible. 1 is also markedly different from the structure exhibited in the related solvent-free tris(amido) lithium magnesiate [LiZn(HMDS)3] which is a monomer where lithium is stabilised by forming intramolecular secondary agostic interactions with trimethylsilyl groups.10
Considering the defining role that aggregation plays in modulating the reactivity of s-block organometallic compounds as well as the fact that crystal structures of many of these species do not correlate with their constitution in solution, we decided to perform 1H NMR diffusion-ordered spectroscopy experiments (1H-DOSY) on 1 in order to assess its aggregation in C6D6 solutions. This methodology can be used to identify different aggregates present in solution as well as to estimate their sizes (which are inversely proportional to their diffusion coefficients, D).11 Seminal contributions by Williard have shown that, using simple organic hydrocarbons as references, accurate hydrodynamic dimensions of several organolithium compounds can be established.11 We chose 1,2,3,4-tetraphenylnaphthalene (TPhN), 1-phenylnaphthalene (PhN) and tetramethylsilane (TMS) as internal standards for this study. The 1H NMR DOSY spectrum of a mixture of 1 with these standards in C6D6 at 27 °C showed that all the components clearly separate in the diffusion dimension according to their increasing D values.‡
A correlation between log D and log FW (FW = molecular weight) of the linear least-squares fit to the internal standards can be established (log D = −0.6341·log FW − 7.4601; r = 0.9923).‡ By interpolating the D value corresponding to the Me3SiCH2 ligand of 1 in this calibration curve, an approximate value of its size (in terms of its FW) can be obtained and therefore a measure of its aggregation in solution.‡1 has an estimated molecular weight of 409 g mol−1. Based on this, Fig. 2 shows some of the possible species present in solution when 1 is dissolved in deuterated benzene, indicating their relative molecular weight and the error associated with each structure with respect to the estimated size predicted for 1 by the DOSY experiment. Analysis of these data suggests that the highly oligomeric constitution of 1 in the solid state is not retained in solution, as the error associated with much smaller aggregates such as a dimer (B) or a trimer (A) is quite high (34 and 56% respectively). In addition, when considering 1 as a simple monomer (D), there is also a significant disparity with its calculated molecular weight and the estimated value obtained in the DOSY studies (−32%). These data can be interpreted as the presence of a monomer/dimer equilibrium in solution, and therefore the constitution of 1 could be represented by [NaMg(CH2SiMe3)3]n, (where n = 1–2). A better correlation is observed for a monomer in which sodium is solvated by a single molecule of deuterated benzene C (−4% error). This type of electrostatic interaction between a neutral arene molecule and an alkali-metal is well known in organometallic chemistry,12 with several examples structurally defined by X-ray crystallography which show that the alkali-metal adopts a perpendicular disposition π engaging with the electron-rich aromatic ring. Supporting this solution scenario C, compound 1 exhibits excellent solubility in arene solvents (toluene or benzene), in contrast to its total insolubility in non-polar solvents such as hexane or cyclohexane where these types of π interactions are not available.
![Possible species of [{NaMg(CH2SiMe3)3}∞] (1) in C6D6 solution with errors (in parentheses) for every consideration with respect to the estimated FW value predicted through DOSY.](/image/article/2011/CC/c0cc02164d/c0cc02164d-f2.gif) | ||
| Fig. 2 Possible species of [{NaMg(CH2SiMe3)3}∞] (1) in C6D6 solution with errors (in parentheses) for every consideration with respect to the estimated FW value predicted through DOSY. | ||
We then explored the co-complexation reaction of two molar equivalents of NaCH2SiMe3 with Mg(CH2SiMe3)2 aiming to prepare the stoichiometric variant [{Na2Mg(CH2SiMe3)4}] (2). Similar as described for 1 a white precipitate was obtained that dissolved on the addition of benzene which led to the formation of colourless crystals. X-ray crystallographic studies revealed the constitution of some of those crystals was [{(NaCH2SiMe3)4}∞] (3) while other crystals were found to be sodium magnesiate 1, suggesting that if the tetraorganomagnesiate 2 is formed in solution, under the conditions of crystallization, it must undergo a redistribution to a mixture of 3 and 1.¶
Featuring a polymeric chain structure built up from {Na(CH2SiMe3)}4 tetramers (Fig. 3), 3 represents a new addition to the exclusive family of unsolvated sodium organometallics structurally defined.3,13 The repeating unit comprises a distorted Na4 tetrahedral core with four alkyl groups, each of them capping a face of the tetrahedron. Two of these alkyl groups also interact with the Na atoms of two neighbouring units [Na2–C2, 2.7872(17) Å], giving rise to the chain arrangement. This interaction induces an asymmetry in the structure of the repeating tetrahedron unit with Na2 raising its coordination number to four whereas Na1 is only bonded to three alkyl groups reflected not only in a variation in the Na–C bond lengths [from 2.5953(19) to 2.8189(18) Å] but also in the non-bonding Na⋯Na distances [varying from 2.9125(14) to 3.3594(10) Å].14 This tetrahedron based motif is also exhibited by classical organometallic reagents3 such as MeLi, MeNa and tBuLi15 and contrasts with the alternating chain structure of Na+ cations and anions of related solvent free [{NaCH(SiMe3)2}∞].13a
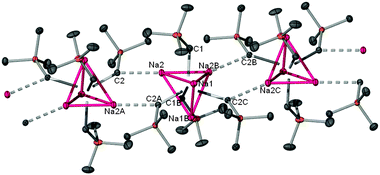 | ||
| Fig. 3 Polymeric chain structure of 3 with hydrogen atoms omitted for clarity. | ||
To conclude, by studying co-complexation reactions of monometallic alkyls, we have uncovered the novel structures of the solvent-free sodium trisalkylmagnesiate 1 and its sodium component 3, which advance understanding of the fascinating correlations existing between structural patterns and reactivities of s-block organometallic compounds.
We thank the EPSRC, the Royal Society, the Carnegie Trust for the Universities of Scotland and the 7th European Community Framework Programme for their generous sponsorship of this research.
Notes and references
- G. Wittig, F. J. Meyer and G. Lange, Justus Liebigs Ann. Chem., 1951, 571, 167 CrossRef CAS.
- H. Yorimitsu and K. Oshima, in The Chemistry of Organomagnesium Compounds, ed. Z. Rappoport and I. Marek, Patai Series, Wiley, Chichester, UK, 2008, pp. 681–715 Search PubMed.
- For a seminal review on structures of organo alkali-metal complexes see: E. Weiss, Angew. Chem., Int. Ed. Engl., 1993, 32, 1501 Search PubMed.
- R. E. Mulvey, Acc. Chem. Res., 2009, 42, 743 CrossRef CAS.
- No similar comparison with Mg(CH2SiMe3)2 is possible since it is completely insoluble in C6D6.
- M. J. Zaworotko, Chem. Commun., 2001, 1 RSC.
- Intramolecular Na–C contacts (bond lengths 2.6566(19) and 2.700(2) Å) are of the same order as that observed in the asymmetric unit (2.6708(19) Å).
- V. L. Blair, A. R. Kennedy, J. Klett and R. E. Mulvey, Chem. Commun., 2008, 5426 RSC.
- (a) K. W. Henderson, A. R. Kennedy, A. E. McKeown and D. Strachan, J. Chem. Soc., Dalton Trans., 2000, 4348 RSC; (b) K. W. Henderson, A. R. Kennedy, L. Macdonald and D. J. MacDougall, Inorg. Chem., 2003, 42, 2839 CrossRef CAS.
- A. R. Kennedy, R. E. Mulvey and R. B. Rowlings, J. Am. Chem. Soc., 1998, 120, 7816 CrossRef CAS.
- D. Li, I. Keresztes, R. Hopson and P. G. Williard, Acc. Chem. Res., 2009, 42, 270 CrossRef CAS.
- G. W. Gokel, S. L. De Wall and E. S. Meadows, Eur. J. Org. Chem., 2000, 2967 CrossRef CAS.
- Selected examples of unsolvated NaR structurally characterised: (a) P. B. Hitchcock, M. F. Lappert, W. P. Leung and T. Shun, J. Chem. Soc., Chem. Commun., 1993, 1386 RSC; (b) M. Niemeyer and P. P. Power, Organometallics, 1997, 16, 3258 CrossRef CAS.
- For a recent example of an asymmetric ether solvated LiR (R = CH2SiMe3) see: T. Tatic, K. Meindl, J. Henn, S. K. Pandey and D. Stalke, Chem. Commun., 2010, 46, 4562 Search PubMed.
- T. Kottke and D. Stalke, Angew. Chem., Int. Ed. Engl., 1993, 32, 580 CrossRef.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Full experimental details and NMR spectra. CCDC 783163–783164. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc02164d |
§ Crystal data for 1: C12H33MgNaSi3, Mr = 309.0, orthorhombic, space groupPbca, a = 16.8522(4), b = 12.7158(4), c = 19.3676(7) Å, V = 4150.3(2) Å3, Z = 8, λ = 0.71073 Å, μ = 0.264 mm−1, T = 150 K; 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 574 reflections measured, 4610 unique, Rint = 0.061; R = 0.036 (F, 2442 data with F2 > 2σ), Rw = 0.071 (F2, all data), GOF = 0.78, 163 refined parameters, final difference map within ±0.3 e Å−3. 574 reflections measured, 4610 unique, Rint = 0.061; R = 0.036 (F, 2442 data with F2 > 2σ), Rw = 0.071 (F2, all data), GOF = 0.78, 163 refined parameters, final difference map within ±0.3 e Å−3. |
| ¶ Crystal data for 3: C4H11NaSi, Mr = 110.21, monoclinic, space groupC2/c, a = 21.481(2), b = 12.4281(7), c = 12.7071(12) Å, V = 2827.1(4) Å3, Z = 16, λ = 0.71073 Å, μ = 0.271 mm−1, T = 123 K; 7086 reflections measured, 3258 unique, Rint = 0.028; R = 0.033 (F, 2273 data with F2 > 2σ), Rw = 0.073 (F2, all data), GOF = 0.928. |
| This journal is © The Royal Society of Chemistry 2011 |