A triangulopalladium cluster consisting of μ3-capping silyl ligands†‡
Felix
Armbruster
a,
Jens
Meyer
a,
Alexander
Baldes
b,
Pascual Oña
Burgos
a,
Ignacio
Fernández
c and
Frank
Breher
*a
aInstitute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstraße 15, 76131 Karlsruhe, Germany. E-mail: breher@kit.de; Fax: +49 721 608 7021; Tel: +49 721 608 4855
bInstitute of Physical Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstraße 15, 76131 Karlsruhe, Germany
cÁrea de Química Orgánica, Universidad de Almería, Ctra. Sacramento s/n, 04120, Almería, Spain
First published on 28th April 2010
Abstract
We report on the utility of multifunctional, κ1Si:κ3S-coordinated tris(methimazolyl)silanide ligands [Si(mtMe)3]− for the stabilisation of a triangulopalladium cluster [Pd3{Si(mtMe)3}2] (3) consisting of very unusual μ3-capping SiR3 donors. Differences to the corresponding platinum chemistry were supported by NMR spectroscopy and DFT calculations.
There is a growing interest in multidentate ligand systems featuring dual functionality.1 Among tripodal shaped ligands, one important subgroup of intramolecularly coordinating, tetradentate ligands is based on chelating donors which are coupled by an additional Lewis-basic coordination site. In most cases, these chelating sets of donors are coordinated to a single metal atom to produce mononuclear complexes with well-defined coordination geometries.
Although relatively rare compared to other well-established systems,2 strongly trans influencing, anionic silanide donors connected to three ancillary donor buttresses have attracted considerable recent interest. Seminal work was published by Hendriksen et al.3 and Stobart et al.4 in the late 1980s on rhodium and iridium complexes consisting of tetradentate [κ1Si:κ3P3]− ligands. Most recently, Peters et al.5 reported inspiring work on 3d transition metal complexes of closely related tris(phosphino)silanides. In extension to our studies on ambidentate tris(pyrazolyl)methanides6 and -silanides7 we became interested in intramolecularly coordinating ligands for the stabilisation of small metal clusters and/or reactive species. Herein, we report the synthesis and characterisation of a trinuclear Pd cluster (3) stabilised by two μ3-capping tris(methimazolyl)silanide ligands [Si(mtMe)3]− (2).
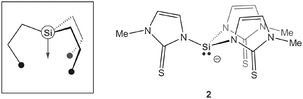
The ligand precursor HSi(mtMe)3 (2H) was synthesised as analytically pure, colourless powder by reacting Me3Si(mtMe) (1) with HSiCl3 in toluene at 70 °C in a closed reaction vessel. 2H is only hardly soluble in most organic solvents, preventing a full NMR spectroscopic characterisation. Nevertheless, the elemental analysis and EI mass spectroscopic data were consistent with the composition HSi(mtMe)3. The Si–H moiety was detected at a characteristic frequency of ν = 2322 cm−1 in the IR spectrum.
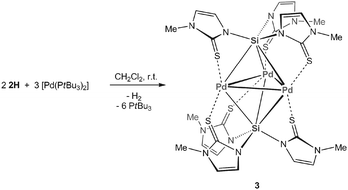
In a second step, 2 equiv. of 2H were reacted with 3 equiv. of the palladium precursor [Pd(PtBu3)2] in CH2Cl2 at room temperature (Scheme 1). The ligand gradually dissolved and the reaction mixture became intensively yellow in colour. Furthermore, the evolution of H2 could be observed (confirmed by 1H NMR). After ca. 2 minutes, stirring was stopped and the reaction vessel was kept at room temperature over night. Air-stable, orange crystals of [Pd3{Si(mtMe)3}2] (3) were isolated by filtration and washed with hexane in analytically pure form in 56% yield. The NMR spectra of 3 were consistent with the structure proposed in Scheme 1. One set of resonances for mtMegroups were observed in the 1H NMR (d6-dmso) in the ratio 1H ∶ 1H ∶ 3H at δH 7.13, 6.26 (each CH) and 3.48 ppm (Me), consistent with κ1Si:κ3S-coordinated ligands (2) and an overall highly symmetric constitution. No signals for SiH or PdH entities could be observed, further supporting the proposed loss of H2 during the complexation. The 29Si NMR chemical shift of δSi 0.56 ppm was detected by acquiring a 1H, 29Si gHMQC 2D experiment (see ESI‡). As expected, two nitrogen chemical shifts were observed by 1H, 15N gHMQC at δN = 157 and 207 ppm. The structural integrity of 3 in solution was supported by pulsed field gradient spin-echo (PGSE) measurements. The hydrodynamic radius (rH = 6.9 Å) and volume (VH = 1375 Å3) of 3 in d6-dmso solutions were obtained by using the experimental diffusion coefficient (D = 1.466·10−10 m2 s−1) together with the Stokes–Einstein equation. VH is in very good agreement with the volume for an individual molecule estimated from X-ray crystallographic studies (VX-ray = 1364 Å3).§
 | ||
| Scheme 1 Synthesis of 3. | ||
The molecular structure of 3 is shown in Fig. 1 and confirms the formation of an overall neutral cluster consisting of a {Pd3Si2} core. Formally, 3 is composed of a [Pd3]2+ triangle, stabilised by two anionic, multifunctional silanide ligands ([Si(mtMe)3]−, 2). The silicon atoms adopt very unusual μ3-capping positions on the triangular faces furnishing an overall trigonal bipyramidal cluster core.8 Each hexacoordinated silicon atom9 is further attached to three ancillary mtMe donors, which are in turn coordinated by the sulfur termini to palladium. Owing to the skew arrangement of the methimazolyl ligands, a very appealing, almost ideal D3 symmetric structure results. The Pd–Pd bond lengths (avg. 2.7311(8) Å) are reasonably comparable to those found for other 44 electron Pd3 clusters containing CO or phosphine ligands.10 The Si–Pd distances of avg. 2.397(2) Å are within the expected range.11
![Displacement ellipsoid plot (30% probability) of [Pd3{Si(mtMe)3}2] (3). For selected distances and angles see ESI.](/image/article/2011/CC/c002435j/c002435j-f1.gif) | ||
| Fig. 1 Displacement ellipsoid plot (30% probability) of [Pd3{Si(mtMe)3}2] (3). For selected distances and angles see ESI.‡ | ||
Compared to the very rich silylplatinum chemistry,11,12 reports on analogous Si/Pd complexes or clusters are fewer in number. Although some dinuclear Pd complexes with (bridging) Si ligands are known,13 compounds of higher nuclearity are relatively rare. Shimada and Tanaka et al. characterised some multinuclear Pd complexes with chelating silyl ligands.14 Only recently, Osakada and co-workers were able to isolate and characterise larger silylene bridged cluster compounds consisting of planar Pd4Si3 scaffolds in their molecular cores.15 As far as we are aware, no trianguloPd cluster with μ3-bridging SiR3 ligands has been reported until now. Most commonly, these types of trinuclear clusters are stabilised by ligands consisting of fundamentally different σ-donor and π-acceptor properties, such as phosphines, carbon monoxide, or isonitriles.16
A density functional theory (DFT) calculation was conducted on 3 at the BP86/def2-TZVP level in D3-symmetry (see ESI‡). As expected for these types of trianguloclusters, the highest occupied molecular orbitals are predominantly Pd-centred. The silicon palladium bonding interactions are of multi-centre type. Natural population analysis (NPA) and population analysis based on occupation numbers (PABOON) revealed that considerable charge density is transferred from the Si atoms to the Pd3 cluster core and the chelating S donors (NPA and PABOON results: Si: +0.239, +1.291; Pd: −0.010, +0.103; S: −0.334, −0.184). This would be in accord with the electropositive nature of the silicon donors.
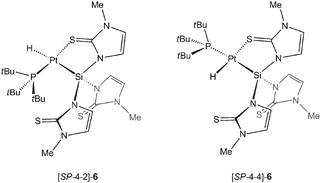
The detailed mechanism for the formation of 3 is unknown. However, it is reasonable to assume that the initial step involves oxidative addition of the Si−H functional group to the zerovalent palladium, possibly under displacement of one PtBu3 and formation of [PdH(PtBu3){Si(mtMe)3}] ([SP-4-2]/[SP-4-4]-4, see Chart 1 for the analogous Pt compound). In a second step, two equiv. of 4 can react with one further equiv. of [Pd(PtBu3)2] to give 3, H2 and free phosphines. In order to identify other products in this reaction or possible intermediates formed en route to 3 we also changed the 2H:[Pd(tBu3)2] stoichiometry to 1 ∶ 1 or 2 ∶ 1. However, except from 3 and non-reacted starting materials, no other products could be isolated from these mixtures. 1H NMR spectroscopic monitoring of freshly prepared 1 ∶ 1 mixtures in CD2Cl2 revealed the initial formation of a transient palladium hydride species (δH −6.35 ppm, presumably [SP-4-2]/[SP-4-4]-4), which disappeared after 30 minutes at room temperature.
 | ||
| Chart 1 | ||
In view of the straightforward access of 3, we speculated that the analogous platinum cluster, [Pt3{Si(mtMe)3}2] (5), could also be prepared using the same synthetic protocol and [Pt(PtBu3)2] instead. Initially, we used a 1 ∶ 1 stoichiometry ratio. 1H NMR spectroscopic monitoring revealed a facile addition of the Si−H functional group of 2H to the zerovalent platinum precursor under displacement of one PtBu3 and formation of [PtH(PtBu3){Si(mtMe)3}] ([SP-4-2]/[SP-4-4]-6). The NMR spectra of the latter at room temperature in CD2Cl2 show averaged signals, one Pt–H moiety with a chemical shift of δH −9.75 ppm (1JPt−H = 1140.1 Hz, 2JP−H = 16.4 Hz), one set of resonances for mtMegroups, and one phosphorus and platinum signal at δP 94.1 and δPt −6331.9 ppm (1JPt−P = 2192.5 Hz), respectively. At 173 K the chemical exchange processes are slowed down with respect to the NMR time scale, and as such two species in a ca. 4 ∶ 1 ratio were observed consistent with the structures for the [SP-4-4]- and [SP-4-2]-isomers of 6 depicted above (see ESI‡). Attempts to isolate these species in pure form failed due to their instability in solution. Most importantly, however, we noticed that addition of [Pt(PtBu3)2] to [SP-4-2]/[SP-4-4]-6 did not lead to 5.
In order to gain further information on the different behaviour of Ptvs.Pd, we carried out additional DFT calculations. We computed the gas phase reaction energies for the formation of the oxidative addition products [MH(PtBu3){Si(mtMe)3}] (M = Pd, Pt; eqn (1)) and noticed that in each case the [SP-4-4]-isomers of these products are more stable by 44 (Pd) and 41 (Pt) kJ mol−1 as compared to the [SP-4-2]-isomers. Overall we found that for Pt ([SP-4-4]-6) this reaction is more exothermic by ΔH0 (0 K) = −71 kJ mol−1 as compared to Pd ([SP-4-4]-4) (−44 kJ mol−1).
| [M(PtBu3)2] + 2H → [MH(PtBu3){Si(mtMe)3}] + PtBu3 | (1) |
| 2[MH(PtBu3){Si(mtMe)3}] + [M(PtBu3)2] → [M3{Si(mtMe)3}2] + H2 + 4PtBu3 | (2) |
Anticipating a hemilabile character of the sulfur donors in 3 we speculated that the triangulocluster could be used as catalyst in organic synthesis. Specifically, we found 3 to effect the Suzuki–Miyaura-type cross-coupling reaction of PhB(OH)2 and PhI under various conditions (Scheme 2). Studies in our lab continue to further explore the catalytic reactivity of small metal clusters of this type and to investigate the coordination chemistry of chelating tris(methimazolyl)silanide ligands towards other transition metals.
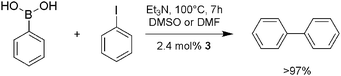 | ||
| Scheme 2 Catalytic studies on 3 (see also ESI‡). | ||
Financial support by the Ministry of Science, Research and the Arts of Baden-Württemberg (Az. 31-655.042-4-1/1) is gratefully acknowledged. IF thanks the Ramón y Cajal program for financial support. We thank Steffen Styra for preparative assistance.
Notes and references
- Review: I. Kuzu, I. Krummenacher, J. Meyer, F. Armbruster and F. Breher, Dalton Trans., 2008, 5836 Search PubMed.
- See e.g. (a) R. R. Schrock, Acc. Chem. Res., 1997, 30, 9 CrossRef CAS; (b) A. G. Blackman, Polyhedron, 2005, 24, 1 CrossRef CAS; (c) F. Mani and L. Sacconi, Comments Inorg. Chem., 1983, 2, 157 CAS.
- D. E. Hendriksen, A. A. Oswald, G. B. Ansell, S. Leta and R. V. Kastrup, Organometallics, 1989, 8, 1153 CrossRef CAS.
- (a) F. L. Joslin and S. R. Stobart, J. Chem. Soc., Chem. Commun., 1989, 504 RSC; (b) R. A. Gossage, G. D. McLennan and S. R. Stobart, Inorg. Chem., 1996, 35, 1729 CrossRef CAS.
- (a) N. P. Mankad, M. T. Whited and J. C. Peters, Angew. Chem., Int. Ed., 2007, 46, 5768 CrossRef CAS; (b) M. T. Whited, N. P. Mankad, Y. Lee, P. F. Oblad and J. C. Peters, Inorg. Chem., 2009, 48, 2507 CrossRef CAS; (c) A. Takaoka, A. Mendiratta and J. C. Peters, Organometallics, 2009, 28, 3744 CrossRef CAS.
- (a) F. Breher, J. Grunenberg, S. C. Lawrence, P. Mountford and H. Rüegger, Angew. Chem., Int. Ed., 2004, 43, 2521 CrossRef CAS; (b) I. Krummenacher, H. Rüegger and F. Breher, Dalton Trans., 2006, 1073 RSC; (c) H. R. Bigmore, J. Meyer, I. Krummenacher, H. Rüegger, E. Clot, P. Mountford and F. Breher, Chem.–Eur. J., 2008, 14, 5918 CrossRef CAS; (d) I. Kuzu, I. Krummenacher, I. J. Hewitt, Y. Lan, V. Mereacre, A. K. Powell, P. Höfer, J. Harmer and F. Breher, Chem.–Eur. J., 2009, 15, 4350 CrossRef CAS; (e) M. G. Cushion, J. Meyer, A. Heath, A. D. Schwarz, I. Fernández, F. Breher and P. Mountford, Organometallics, 2010, 29, 1174 CrossRef CAS; (f) H. R. Bigmore, S. R. Dubberley, M. Kranenburg, S. C. Lawrence, A. J. Sealey, J. D. Selby, M. A. Zuideveld, A. R. Cowley and P. Mountford, Chem. Commun., 2006, 436 RSC; (g) S. C. Lawrence, M. E. G. Skinner, J. C. Green and P. Mountford, Chem. Commun., 2001, 705 RSC; (h) P. K. Byers, N. Carr and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1990, 3701 RSC; (i) P. K. Byers and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1991, 93 RSC.
- (a) F. Armbruster, I. Fernández and F. Breher, Dalton Trans., 2009, 5612 RSC; (b) I. Fernández, P. O. Burgos, F. Armbruster, I. Krummenacher and F. Breher, Chem. Commun., 2009, 2586 RSC.
- For related structural motifs in Pt/Sn clusters see: (a) M. C. Jennings, G. Schoettel, S. Roy, R. J. Puddephatt, G. Douglas, L. Manojlović-Muir and K. W. Muir, Organometallics, 1991, 10, 580 CrossRef CAS; (b) R. V. Lindsey, G. W. Parshall and U. G. Stolberg, Inorg. Chem., 1966, 5, 109 CrossRef CAS; (c) L. J. Guggenberger, Chem. Commun. (London), 1968, 512 RSC.
- Hexacoordinated silicon atoms attached to three transition metals and three non-metals have, to the best of our knowledge, not been described so far. For some alkali metal derivatives see: (a) C. Krempner, M. H. Chisholm and J. Gallucci, Angew. Chem., Int. Ed., 2008, 47, 410 CrossRef CAS; (b) D. M. Jenkins, W. Teng, U. Englich, D. Stone and K. Ruhlandt-Senge, Organometallics, 2001, 20, 4600 CrossRef CAS; (c) A. Sekiguchi, M. Nanjo, C. Kabuto and H. Sakurai, Organometallics, 1995, 14, 2630 CrossRef CAS.
- (a) A. D. Burrows and D. M. P. Mingos, Transition Met. Chem., 1993, 18, 129 CrossRef CAS; (b) D. Kovala-Demertzi, in Metal Clusters in Chemistry, ed. P. Braunstein, L. A. Oro and P. R. Raithby, Wiley-VCH, Weinheim, 1999, vol. 1, p. 399 Search PubMed.
- J. Y. Corey and J. Braddock-Wilking, Chem. Rev., 1999, 99, 175 CrossRef CAS.
- See e.g. (a) R. Waterman, P. G. Hayes and T. D. Tilley, Acc. Chem. Res., 2007, 40, 712 CrossRef CAS; (b) U. Schubert, Transition Met. Chem., 1991, 16, 136 CrossRef CAS; (c) H. Ogino and H. Tobita, Adv. Organomet. Chem., 1998, 42, 223 CAS.
- See e.g. (a) A. Fürstner, H. Krause and C. W. Lehmann, Chem. Commun., 2001, 2372 RSC; (b) W. A. Herrmann, P. Härter, C. W. K. Gstöttmayr, F. Bielert, N. Seeboth and P. Sirsch, J. Organomet. Chem., 2002, 649, 141 CrossRef CAS; (c) M. Suginome, Y. Kato, N. Takeda, H. Oike and Y. Ito, Organometallics, 1998, 17, 495 CrossRef CAS; (d) Y.-J. Kim, S.-C. Lee, J.-I. Park, K. Osakada, J.-C. Choi and T. Yamamoto, Organometallics, 1998, 17, 4929 CrossRef CAS; (e) Y.-J. Kim, S.-C. Lee, J.-I. Park, K. Osakada, J.-C. Choi and T. Yamamoto, J. Chem. Soc., Dalton Trans., 2000, 417 RSC.
- (a) W. Chen, S. Shimada and M. Tanaka, Science, 2002, 295, 308 CrossRef CAS; (b) S. Shimada, Y.-H. Li, Y.-K. Choe, M. Tanaka, M. Bao and T. Uchimaru, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7758 CrossRef CAS; (c) S. Shimada and M. Tanaka, Coord. Chem. Rev., 2006, 250, 991 CrossRef CAS.
- T. Yamada, A. Mawatari, M. Tanabe, K. Osakada and T. Tanase, Angew. Chem., Int. Ed., 2009, 48, 568 CrossRef CAS.
- (a) R. J. Puddephatt, L. Manojlovic-Muir and K. W. Muir, Polyhedron, 1990, 9, 2767 CrossRef CAS; (b) D. Imhof and L. M. Venanzi, Chem. Soc. Rev., 1994, 23, 185 RSC; (c) A. D. Burrows and D. M. P. Mingos, Coord. Chem. Rev., 1996, 154, 19 CrossRef CAS.
- We have no experimental evidence for a one-step formation of the triangulocluster according to eqn (2). This hypothetical process was calculated only in order to estimate the differences between both cases.
- V. V. Grushin, Chem. Rev., 1996, 96, 2011 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and NMR spectra. CCDC 764799. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c002435j |
§ Crystal data of 3: C24H30N12Pd3S6Si2·4(CH2Cl2), 1394.04 g mol−1, orthorhombic, P212121; a = 12.595(3), b = 19.333(4), c = 19.099(4) Å, V = 4848(2) Å3, T = 150 K, Z = 4, ρcalcd = 1.910 Mg m−3, μ(MoKα) = 1.889 mm−1, crystal dimensions: 0.25 × 0.24 × 0.24 mm, 2Θmax = 52°, 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 795 reflections, 9444 independent (Rint = 0.0761), 566 parameters, 21 restraints, R1 = 0.0449 and wR2 (all data) = 0.1985, Flack χ parameter −0.03(3), largest final difference peak/hole +0.889/−0.930 e Å−3. CCDC 764799. 795 reflections, 9444 independent (Rint = 0.0761), 566 parameters, 21 restraints, R1 = 0.0449 and wR2 (all data) = 0.1985, Flack χ parameter −0.03(3), largest final difference peak/hole +0.889/−0.930 e Å−3. CCDC 764799. |
| This journal is © The Royal Society of Chemistry 2011 |