A comparative study on dissolution rate of sintered (Th–U)O2 pellets in nitric acid by microwave and conventional heating
G.
Singh
a,
R. K.
Singhal
*ab,
R. K.
Malav
a,
A.
Fulzele
a,
A.
Prakash
a,
Md.
Afzal
a and
J. P.
Panakkal
a
aAdvanced Fuel Fabrication Facility, BARC, Tarapur, 401502, India. E-mail: rsinghal@barc.gov.in
bAnalytical Chemistry Division BARC, Mumbai, 400084, India
First published on 28th January 2011
Abstract
ThO2 with approximately 3% 233UO2 is the proposed fuel for the Advanced Heavy Water Reactor (AHWR) and characterized as a mixed oxide (MOX) fuel. Due to the existence of a single oxidation state, quantitative dissolution of MOX samples like (Th–U)O2 and (Th–Pu)O2 is a challenging task for any analytical chemist. However, dissolution is a pre-requisite for complete characterization of the fuel. The present paper describes a detailed study carried out on the comparison of dissolution of sintered (Th–U)O2 mixed oxide pellets, in 16 M HNO3 using microwave and conventional heating techniques, under reflux conditions. The study was carried out with variation of parameters such as concentration of HF and UO2 substitution in the MOX solid solution. The concentration of dissolved U and Th was determined by modified Davies and Gray potentiometric titration and Th-EDTA complexometric titration using xylenol orange as the titration end point indicator respectively. Experimental results clearly indicate that the microwave heating assisted dissolution rate is 2 to 3 times higher than the conventional infrared heating assisted dissolution rate, for sintered pellets. The concentration of HF is a critical parameter, an excess leads to precipitation of insoluble ThF4. The concentration of HF was optimized to 0.025 M. Experimental results also clearly show that at this concentration of HF in 16 M HNO3, a higher substitution of UO2 in MOX solid solution also facilitates the dissolution process. Different trace metal impurities were determined in the MOX samples after dissolving by microwave and infra red heating techniques and it was observed that the results are comparable.
1.0 Introduction
The Advanced Fuel Fabrication Facility (AFFF), Tarapur, is fabricating various types of mixed oxide fuels for different Indian reactors. ThO2 containing around 4% 233UO2 is the proposed fuel for the Advanced Heavy Water Reactor (AHWR).1,2 The fabrication of 233Th-U MOX fuel is difficult as it usually contains daughter products of 232U (T½ = 68.9 years) namely 212Bi and 208Tl which emit strong gamma radiations 0.7–1.8 MeV and 2.6 MeV respectively. So the fabrication of the above fuel requires operation in a shielded glove box to protect operators from exposure. Conventionally, the above fuel is fabricated by a powder metallurgy route which has been replaced by the Coated Agglomerate Pelletization (CAP) process. Under this process, direct blending of 233UO2 and ThO2 powders was done.2The chemical characterization of nuclear fuels plays a crucial role in improving the performance of the fuel in the reactor. The various chemical parameters required to be determined are heavy metal content, trace metallic and non-metallic impurities, O/M [oxygen/metal] mole ratio etc. Specification like heavy metal content is important from a fissile content point of view. The titrimetric methods are best rated for the heavy metal determination due to their excellent precision. Thorium is determined by Th-EDTA complexometric titration3,4 followed by either visual end point detection or by spectrophotometry. A controlled potential coulometric method has been reported in the literature for determination of thorium, using mercury as the working electrode where a precision better than ±0.2% (RSD) has been achieved for thorium in the range of 4 to 8 mg.4 Quantitative determination of uranium is done by a modified Davies and Gray redox titrimetric method5 followed by potentiometric end point determination.
Numerous experimental methods have been reported in the literature for measurement of oxygen to metal mole ratio and they are broadly classified based on dry and wet chemical methods.6 Analytical methods routinely being used for O/M determination are the gas equilibration method, coulometric method and oxidation–reduction method.7–9
To maintain neutron economy and fuel density it is necessary to have control on trace constituents in the fuel. For common metallic impurities, various analytical techniques like direct current arc emission spectroscopy (DC-Arc-AES), X-ray fluorescence (XRF), ion selective electrode (ISE) and atomic absorption spectroscopy have been employed.10,11 DC-Arc-AES is a sensitive technique but its precision is poor.12ISE is a single element technique and its detection limits are high.13 The sensitivity of XRF is poor for elements of lower atomic number.14Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) has emerged as a very powerful technique due to its sensitivity, high linear dynamic range and precision.15
In a nuclear fuel fabrication facilities like AFFF(Advance Fuel Fabrication Facilities), the total analysis time is very important so that the fuel can be supplied as per schedule. As both these specifications (heavy metal content and metallic impurities) require sample in the solution form, analytical chemists are looking forward for new dissolution methodologies. Microwave heating is observed with a number of unique advantages compared to conventional infra red heating.16–19 These include non-contact heating, rapid heating, reduced processing costs, material selective heating, volumetric heating, reverse thermal gradient, uniform heating, quick start up and stopping. Microwave dissolution due to its complete heating promises, has an edge over the conventional mode of heating. It has emerged as a powerful heating technique and has been applied in nuclear chemistry and engineering over the past decades.20,21 Attempts have already been made earlier to see the comparative dissolution using microwave heating with UO2 powder and pellets.22,23 In the present paper, a detailed study has been reported on the dissolution of (Th–U)O2 mixed oxide fuel in view of the requirements in an analytical laboratory.
In the present work dissolution of sintered (Th–U)O2 MOX pellets in 16 M HNO3 using microwave and conventional electrical heating were carried out. A comparison on the basis of percent dissolution versus time curves has been made. Efforts were made to optimize the chemical parameters during dissolution like HF and HNO3 concentration for faster dissolution of ThO2 and ThO2 based MOX fuel. A lesser use of HF will reduce the chances of corrosive deterioration of glove boxes as well as components of analytical instruments. The dissolution data of ThO2 and ThO2 based MOX fuel is important to enhance the production rate at the front end (fabrication) and back end of nuclear fuel cycle (reprocessing).
2.0 Experimental
2.1 Materials
Sintered pellets of two compositions namely ThO2-3.75%UO2 and ThO2-5%UO2 were taken. Densities achieved after sintering in air at 1400 °C were 95% of theoretical density (TD). Theoretical densities for UO2 and ThO2 are 10.97 g cm−3 and 10.00 g cm−3 respectively.27,282.2 Reagents
Reagents of highest purity grade were used: 65% HNO3 (Suprapure, Merck) and deionized water from Millipore RiOs™. The stock solution of uranium and thorium was prepared from urinyl nitrate and thorium nitrate. Stock solutions for different matrix elements were prepared by using E-Merck, India standard solutions having a concentration of 1000 μg mL−1 in 1% HNO3.2.3 Apparatus
Conventional heating was carried out in a resistance heater (500 W) with a temperature controller (± 1 °C) and microwave heating in a 700 W stainless steel indigenous microwave oven adjustable with an increment of 1% in the span of 0% to 100%. Quartz flasks fitted with a water condenser were used for dissolution experiments carried out under reflux conditions. The pH of the samples were measured by a digital pH meter (make Thermo scientific) with an accuracy of (±0.002). Five different sets of experiments were carried out for both the dissolution process. For Th determination two concentrations of titrant (EDTA) were used namely 0.01 M and 0.001 M. Similarly for U determination two concentrations of titrant K2Cr2O7 were taken. Pre-weigh polypropylene burettes having 10 mL capacity with narrow bores were used for titration. While performing the titration the burette was squeezed with fingers such that the solution flows out freely in the form of drops. All the titrations were carried out on the weight basis.Determination of oxygen to metal [O/(Th + U)] mole ratio was determined by the oxidation–reduction method9 in a thermogravimetric analyser [TGA-Setsys evaluation16,18 coupled with an evolve gas analyser [EGA-Quadstar 32 Bit, QMS-200]. The sintered pellet was crushed into chips and a weighed chip was taken in an alumina crucible and kept in the graphite furnace of a thermogravimetric analyzer. It was subjected to a programmed heating profile. Before starting the experiment, the chamber was evacuated using a rotary pump until a pressure of 20 mbar is achieved. In the oxidation step, the sample was heated in the presence of ultra high pure oxygen gas up to 800 °C at a rate of 5 °C min−1 and kept for two hours at that temperature. It was allowed to cool up to room temperature at a rate 10 °C min−1. In the reduction step the oxidized sample was reduced under Ar-8% H2 atmosphere at the same heating profile as that of the oxidation step.
2.4 Dissolution of sintered MOX pellets
(Th-3.75%U)O2 and (Th-5%U)O2 pellets fabricated by CAP process were dissolved in 60 mL 16 M HNO3/HF mixtures at a boiling temperature (∼120 °C) by conventional and microwave heating under reflux conditions. Samples were withdrawn at regular intervals (two at an instant) of time until the 100% dissolution of a pellet.2.5 Determination of uranium (U) and thorium (Th) content
2.6 Determination of trace metallic impurities
Trace metallic impurities in (Th–U)O2 MOX pellets were determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) after separation of the major matrix of thorium and uranium.15,29 A sintered ThO2-3.75%UO2 MOX pellet (∼6g) was crushed into chips and two samples (each ∼3 g) were dissolved in the microwave digestion system and conventional heating under reflux conditions in 16 M HNO3/HF mixtures separately. The solutions were evaporated to dryness twice to remove fluoride and final volumes (∼20 ml) were made up in 5 M HNO3.
Thorium and uranium were separated by solvent extraction using (40% tributyl phosphates {TBP}–0.2 M trioctylphosphide oxide {TOPO})/CCl4/HNO3. These were separated by giving first four contacts with 40% TBP in CCl4 and the last two contacts with 0.2 M TOPO in CCl4. In the first four contacts the organic to aqueous ratio was maintained at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 while in the last two contacts it was 1:1. The aqueous phase obtained after solvent extraction contains common metallic impurities along with some residual thorium and uranium.
:1 while in the last two contacts it was 1:1. The aqueous phase obtained after solvent extraction contains common metallic impurities along with some residual thorium and uranium.
Both these samples for various metallic impurities were analyzed by inductive coupled plasma atomic emission spectroscopy (ICP-AES, Model JY ULTIMA 2 CHR), having both polychromator and monochromator. The polychromator uses thirty six photomultiplier tubes as detectors which correspond to a specific element. But, in the present work, the monochromator was exclusively used due to its better sensitivity. The entire facility is specially modified to handle radioactive material. The Normal Analytical Zone Multi-WAV acquisition mode provides complete sample fingerprinting of 75 elements in less than 30 s. The details of various optimized parameters are given in Table 1. Calibration of the instrument was carried out by using a standard solution of different elements from Fluka/Aldrich. The main operating conditions of the instruments are given in the Table 1.
| Monochromator | |
| Mounting | Czerny-Turner, focal length 1 m |
| Grating and wavelength range | Holographic, 3600 grooves mm−1 |
| Spectral resolution | Less than 12 pm at 190 nm and less than 11 pm at 400 nm and 21 pm at 190 nm. |
| Detectors | PMT |
| Rf generator | Solid state with water cooling |
| Frequency | 40.68 MHz |
| Power output | 0.7–1.0 kW |
| Observation | Radial |
| Nebulizer | Cross flow |
| Type of spray chamber | Scott |
| Plasma torch | Fully demountable torch |
| Injector tube diameter of torch | 3 mm |
| Pump | Peristaltic, three channels, twelve-roller |
| Operating conditions | |
| Outer argon flow rate (L min−1) | 12 |
| Sheath gas flow rate (L min−1) | 0.2 |
| Liquid uptake rate (mL min−1) | 1.8 |
| Nebulizer pressure (bar) | 3.2 |
| Nitrogen flow rate: | λ < 200 nm, λ < 400 and λ > 400 optics was purged by 6 L min−1, 4 L min−1 and 0.5 L min−1 |
| Incident power (kW) | 1 |
3.0 Results and discussion
Various physiochemical characteristics of the MOX fuel pellets were measured before dissolution. The density of the MOX pellets is 95% to theoretical density (TD) of the pellets of UO2 (10.97 g cm−3) and ThO2 10.00 (g cm−3). Fig. 1 and 2 shows the thermogram under oxidation and reduction conditions respectively along with the heating profile. The O/U + Th mole ratio was calculated using the following expressionwhere M is the molecular weight of the (Th–U)O2 sample, Wi and Wf are initial (before oxidation) and final (after reduction) weights of the sample. The O/U + Th ratio for the above sample was found to be 2.007 ± 0.002.

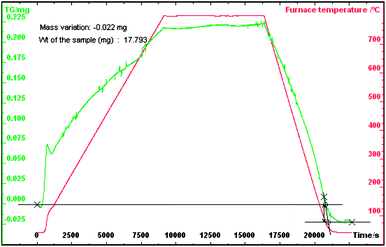 | ||
| Fig. 1 Thermogram of sintered ThO2-5% UO2 MOX in an O2 atmosphere. | ||
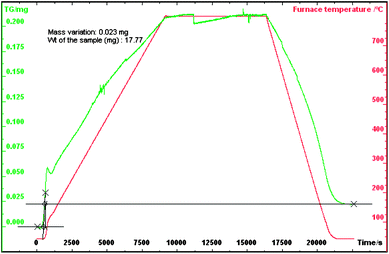 | ||
| Fig. 2 Thermogram of oxidized sintered ThO2-5% UO2 MOX in an Ar-8%H2 atmosphere. | ||
3.1 Influence of concentration of HF on dissolution
ThO2 and ThO2 based MOX fuels are relatively inert and refractory as thorium has only one oxidation state i.e +4, so oxidative–reductive methods are meaningless. Thermodynamics is not favorable for dissolution of ThO2 in pure nitric acid and fluoride ion is required to catalyze the dissolution of pure ThO2 and (Th–U)O2 MOX pellets with ThO2 up to 60%.24–26 However the concentration of fluoride needs optimization due to corrosive characteristics of fluoride. Fig. 3 and 4 shows comparative dissolution under conventional reflux conditions at 120 °C and microwave heating respectively for dissolution of (Th-3.75% U)O2 sintered pellet in 0.025 M and 0.05 M HF in 16 M HNO3![Dissolution of (Th-3.75% U)O2 sintered pellet in 0.025 M and 0.05 M HF in 16 M HNO3 by [A] conventional heating and [B] microwave under reflux conditions at 120 °C.](/image/article/2011/AY/c0ay00630k/c0ay00630k-f3.gif) | ||
| Fig. 3 Dissolution of (Th-3.75% U)O2 sintered pellet in 0.025 M and 0.05 M HF in 16 M HNO3 by [A] conventional heating and [B] microwave under reflux conditions at 120 °C. | ||
![Dissolution of (Th-5% U)O2 sintered pellet in 0.025 M and 0.05 M HF in 16 M HNO3 by [A] conventional heating and [B] Microwave heating under reflux conditions at 120 °C.](/image/article/2011/AY/c0ay00630k/c0ay00630k-f4.gif) | ||
| Fig. 4 Dissolution of (Th-5% U)O2 sintered pellet in 0.025 M and 0.05 M HF in 16 M HNO3 by [A] conventional heating and [B] Microwave heating under reflux conditions at 120 °C. | ||
The experiments were also performed on sintered (Th-5%U)O2 pellets and a variation of dissolution with time is shown in Fig. 3 and 4. It is evident from the figures that for a particular MOX solid solution composition, the higher the concentration of HF, the faster the dissolution process. As discussed before, the concentration of HF is a critical parameter; the use of a concentration higher than 0.05 M may lead to precipitation of insoluble ThF4. Therefore it is advisable to use an optimized concentration of HFi.e. 0.025 M.
From Fig. 3 and 4 its also evident that for a given concentration of HF in 16 M HNO3, the time of dissolution decreases as the percentage of UO2 in (Th–U)O2 MOX solid solution increased. This is attributed to UO2 as it can extremely absorb microwave energy, rapidly leading to a high temperature. The nitric acid and uranium dioxide are both polar molecules with a high value of dielectric loss factor and can absorb microwaves significantly.30,31 This result may also be interpreted by the oxidation of U(IV) in contact with HNO3 (dissolvent) into U(VI) which increases the dissolution rate of the matrix or due to the presence of more uranium in the lattice which is easily attacked by HNO3 creating lattice defects that help the dissolution process.
3.2 Determination of dissolved uranium and thorium content
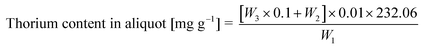 | (1) |
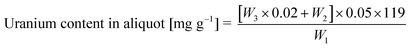 | (2) |
| S. No | Sample | Mode of Dissolution | Number of Repetition | Time for 100% dissolution (h) | U (%) | Th (%) |
|---|---|---|---|---|---|---|
| 1 | ThO2-3.75%UO2 | Conventional | 5 | 12–14 | (3.23 ± 0.02) | (83.62 ± 0.47) |
| 2 | ThO2-3.75%UO2 | Microwave | 5 | 5–7 | (3.19 ± 0.04) | (83.51 ± 0.41) |
| 3 | ThO2-5%UO2 | Conventional | 5 | 12–14 | (4.43 ± 0.04) | (82.92 ± 0.37) |
| 4 | ThO2-5%UO2 | Microwave | 5 | 5–6 | (4.54 ± 0.04) | (82.84 ± 0.39) |
3.3 Quality assurance of the measurements of U and Th
There is no standard reference material available for MOX fuel, therefore utmost attention is given for precision. A precision of better than 0.20% at the 95% uncertainty level (n = 5) has been obtained.3.4 Comparisons of microwave with conventional heating
In both the composition of Th–U and at different concentration of HF, microwave assisted dissolution is always faster by 30–40% compare to conventional heating. Comparative dissolution under microwave and conventional heating of (Th-3.75%U)O2 and (Th-5%U)O2 having similar HF concentration (0.025 M) is shown in Fig. 3 and 4 whereas at 0.05 M HF is shown in Fig. 3 and 4. As expected an increase in % dissolution was observed due to the presence of uranium in the solid solution, at any instant, for a given HF concentration in 16 M HNO3. So, the time of dissolution decreases as the percentage of U in (Th–U) MOX solid solution increased. This result was interpreted by the oxidation of U(IV) in contact with HNO3 (dissolvent) into U(VI) which increases the dissolution rate of the matrix.3.5 Influence of mode of heating
For conventional heating, the energy must be conducted through the walls of the vessel containing the solution and is restricted to surface only whereas microwave heating results in volumetric heating of pellet and dissolvent. Nitric acid and UO2 can extremely absorb microwave energy,11 resulting in a rapid high temperature. The microwave heating assisted dissolution rate is 2 to 3 times higher than the conventional assisted dissolution rate, for sintered pellets under experimental conditions. So it has been demonstrated that MH can remarkably promote the dissolution process as compared to conventional heating. In the case of microwave associated digestion at temperature 120 °C under closed vessels, microwave decomposition allows the reagents to be heated above their atmospheric boiling points. The higher temperatures achieved in the closed system give the microwave decomposition an advantage over the conventional heating under reflux conditions at 120 °C, as described by the Arrhenius equation:| dln k/dt = Ea/RT2 |
Integration of this equation gives:
| ln k2/k1 = Ea/2.303R(1/T1 − 1/T2) |
In this expression k1 and k2 are rate constants for the reaction of interest at T1 and T2 respectively, Ea is the activation energy and R is the ideal gas constant. These equation show that the reaction rate increases exponential with increasing temperature. This translate into approximately 100 fold decrease in the time required to carry out digestion at 120 °C when compared to conventional heating.32 In addition, because the mineral acid converts the microwave energy into heat almost instantaneously rapid heating is achieved, further decreasing the reaction time.
3.6 Profile of trace metallic impurities
Comparative determination of lanthanide and actinides and alkaline earths and transition metals in ThO2-3.75% UO2 fuel under microwave and conventional dissolution is shown in Fig. 5 and 6 respectively. These impurities were analysed after extraction of the major matrix uranium and thorium using TBP in CCl4 and 0.2 M TOPO in CCl4. From these figures its clear that there is no loss of trace metal weather dissolution is carried out by microwave or conventional heating. Minor variation is mainly statistical in nature. In order to ascertain any loss of trace metals during dissolution process, samples were spiked with a known concentration of trace metal impurities. In both the dissolution process i.e. conventional and microwave, recoveries are in the range of 97–99%.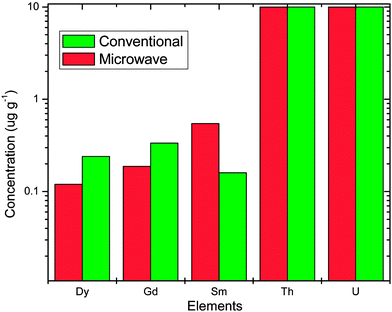 | ||
| Fig. 5 Variation in concentration of lanthanide and actinide in dissolution carried out by microwave and conventional heating. | ||
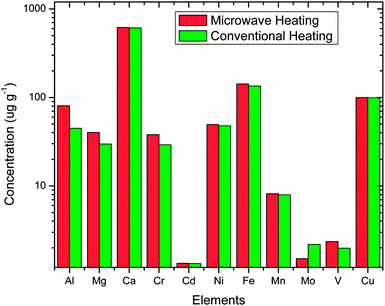 | ||
| Fig. 6 Variation in concentration of transition and alkali metals in dissolution carried out by microwave and conventional heating. | ||
4.0 Conclusions
Microwave heating being volumetric, dissolution of sintered (Th–U)O2 pellets is two–three times faster than conventional heating. So, using the microwave dissolution technique, the analysis time can be reduced significantly. As economy of time is very important in a fuel fabrication facility, the analytical time for the complete fuel characterization should be as small as possible to supply fuel as per schedule. Dissolution experiments shows that 0.025 M HF coupled with microwave heating leads to reasonably faster dissolution which leads to less corrosive deterioration of glove boxes as well as components of analytical instruments. As well as substitution of UO2 in MOX solid solution assists in the dissolution process.Acknowledgements
The authors are grateful to Shri H. S. Kamath, Director Nuclear Fuels Group, for his keen interest and constant encouragement during the course of this workReferences
- R. K. Sinha and A. Kakodkar, Nucl. Eng. Des., 2006, 236, 683–700 CrossRef CAS.
- T. R. G. Kutty, K. B. Khan, P. S. Somayajulu, A. K. Sengupta, J. P. Panakkal, Arun Kumar and H. S. Kamath, J. Nucl. Mater., 2008, 373, 299–308 CrossRef CAS.
- A. I. Vogel, Text book of Quantitative Inorganic Analysis, 3rd edition, Logmans, Green and Co. Ltd, London 2003, p. 442 Search PubMed.
- S. G. Talnikar and R. G. Bhogale, J. Radioanal. Nucl. Chem., 1990, 141(2), 401–405 CAS.
- W. Davies and W. Gray, Talanta, 1203, 1964, 11 Search PubMed.
- J. Robin, K. Chidester and M. Thompson, Report LA-UR-00–5805.
- C. E. Mcneilly and T. D. Chikalla, J. Nucl. Mater., 1971, 39, 77 CrossRef CAS.
- C. F. Metz, J. W. Dahlby and G. R. Waterbury, IAEA-SM-149/33, Vienna, ( 1972) 35.
- E. A. Schaefer and J. O. Hibbits, Anal. Chem., 1969, 41(2), 254 CrossRef CAS.
- Annual book of ASTM standards ( 1999) Vol. 12.01 Nucl. energy (I) C698, pp. 46–75 Search PubMed.
- V. Natrajan, B. A. Dhawale, B. Rajeshwari, N. S. Hon, S. K. Tulsidas, N. K. Porwal, S. V. Godbole and V. K. Manchanda, Spectrochim. Acta, Part B, 2008, 63, 817–819 CrossRef.
- Subramanian R. Venkata, Anal. Lett., 1981, 14, 731.
- C. A. Lucy, J. Chromatogr., A, 1998, 804, 3–15 CrossRef CAS.
- N. L. Mishra, K. D. Mudher Singh, V. C. Adya, B. Rajeshwari and V. Venugopal, Spectrochim. Acta, Part B, 2008, 60, 28–34.
- M. Goaplkrishnan, K. Radhakrishnan, P. S. Dhami, V. T. Kulkarni, M. V. Joshi, A. B. Patwardhan, A. Ramanujam and J. N. Mathur, Talanta, 1997, 44, 169–176 CrossRef CAS.
- D. L. Link, H. M. Kingston, G. L. Havirilla and L. P. Colletti, Anal. Chem., 2001, 74(5), 1165–1170.
- K. E. Haque, Int. J. Miner. Process., 1999, 57, 1–24 CrossRef CAS.
- W. H. Sutton, Am. Ceram. Soc. Bull., 1989, 68(2), 376–386.
- E. T. Thostenson and T. W. Chow, Composites, Part A, 1999, 30, 1055–1071 CrossRef.
- Y. Jhao and J. Chen, Prog. Nucl. Energy, 2008, 50, 1–6 CrossRef.
- G. K. Mallik, R. K. Malav, J. P. Panakkal and H. S. Kamath, Int. J. Nucl. Energy Sci. Tec., 2005, 1(2/3), 16–21 Search PubMed.
- Y. Jhao and J. Chen, Radiochim. Acta, 2008, 96, 467–471 CrossRef.
- Y. Jhao and J. Chen, J. Nucl. Mater., 2008, 373, 53–58 CrossRef.
- W. D. Bond, ORNL-2519, Oak Ridge National Laboratory, 1958. (Dissolution of sintered Thorium-Uranium Oxide fuel in HNO3-HF solutions) Search PubMed.
- H. D. Greiling and K. H. Lieser, Radiochim. Acta, 1984, 36, 79–89.
- E. Zimmer and E. Merz, J. Nucl. Mater., 1984, 124, 64–67 CrossRef CAS.
- T. R. G. Kutty, P. S. Somayajulu, K. B. Khan, Arun Kumar and H. S. Kamath, J. Nucl. Mater., 2009, 389, 351–358 CrossRef CAS.
- The Chemistry of Actinide and Trans-actinide Elements, 3rd Ed., vol. 1, Springer, 2006 Search PubMed.
- A. Kelkar, A. Prakash, Mohd. Afzal, J. P. Panakkal and H. S. Kamath, J. Radioanal. Nucl. Chem., 2010, 284, 443–449 CrossRef CAS.
- C. E. Holocombe, Am. Ceram. Soc. Bull., 1983, 62, 1388.
- P. A. Hass, Am. Ceram. Soc. Bull., 1979, 58, 873.
- R. K. Singhal Manisha Venkatesh, H. Basu, U. Narayanan, A. V. R. Reddy and T. Mukherjee, J. Radioanal. Nucl. Chem., 2010, 285, 353–358 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |