An ultra sensitive saccharides detection assay using carboxyl functionalized chitosan containing Gd2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :Eu3+ nanoparticles probe
:Eu3+ nanoparticles probe
Ashutosh
Tiwari
*ab,
Dohiko
Terada
a,
Prashant K.
Sharma
c,
Vyom
Parashar
c,
Chiaki
Yoshikawa
d,
Avinash C.
Pandey
c and
Hisatoshi
Kobayashi
*ae
aBiomaterials Center, National Institute for Materials Science, 1-2-1, Sengen, Tsukuba, Ibaraki 305 0047, Japan. E-mail: tiwari.ashutosh@nims.go.jp; ashunpl@gmail.com
bJSPS, Sumitomo-Ichibancho Bldg. 6 Ichibancho, Chiyoda-ku, Tokyo 1028471, Japan
cNanophosphor Application Centre, Faculty of Science, University of Allahabads, Allahabad, 211002, India
dInternational Center for Materials Nanoarchitectonics, 1-2-1, Sengen, Tsukuba, Ibaraki 305 0047, Japan
eJST CREST, Kawaguchi, Saitama 3320012, Japan. E-mail: kobayashi.hisatoshi@nims.go.jp; Fax: (+81) 29-859-2247; Tel: (+81) 29-860-4495
First published on 15th December 2010
Abstract
A novel saccharides detection assay based on covalent immobilization of amino phenyl boronic acid (APBA) in thin films of carboxyl functionalized chitosan (HOOC-chitosan) containing <5 nm Gd2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Eu3+ nanoparticles at a platinum disc electrode was developed. The resulting HOOC-chitosan/Gd2O3:Eu3+ nanocomposite film exhibited excellent electrochemical response to changes in the pKa values of boronate esters yielded from different vicinal diols of sugars. The covalent interaction of APBA onto the HOOC-chitosan/Gd2O3:Eu3+ Pt-disc electrode was characterized with FT-IR, SEM, contact angle and cyclic voltammetry, whereas Gd2O3:Eu3+ nanoparticles and HOOC-chitosan/Gd2O3:Eu3+ nanocomposite was identified using XRD, EDX and TEM. A wide linear response was measured to boronate esters ranging from 25 nM to 13.5 μM (r2 = 0.963) with good reproducibility. The excellent electrochemical activity of the assay might be attributed to the synergistic effects of the balanced de-/protonated HOOC-chitosan, APBA and Gd2O3:Eu3+ nanoparticles. With APBA as a model, the HOOC-chitosan/Gd2O3:Eu3+ nanocomposite-modified Pt-electrode was constructed through a simple drop coating method. The resulting assay exhibited a good potentiometric response to different saccharides including glucose, and could be a promising application for the precise electrochemical detection of vicinal diols of specific sugars for clinical diagnostics, medicine validation, bioscience research and food analysis.
:Eu3+ nanoparticles at a platinum disc electrode was developed. The resulting HOOC-chitosan/Gd2O3:Eu3+ nanocomposite film exhibited excellent electrochemical response to changes in the pKa values of boronate esters yielded from different vicinal diols of sugars. The covalent interaction of APBA onto the HOOC-chitosan/Gd2O3:Eu3+ Pt-disc electrode was characterized with FT-IR, SEM, contact angle and cyclic voltammetry, whereas Gd2O3:Eu3+ nanoparticles and HOOC-chitosan/Gd2O3:Eu3+ nanocomposite was identified using XRD, EDX and TEM. A wide linear response was measured to boronate esters ranging from 25 nM to 13.5 μM (r2 = 0.963) with good reproducibility. The excellent electrochemical activity of the assay might be attributed to the synergistic effects of the balanced de-/protonated HOOC-chitosan, APBA and Gd2O3:Eu3+ nanoparticles. With APBA as a model, the HOOC-chitosan/Gd2O3:Eu3+ nanocomposite-modified Pt-electrode was constructed through a simple drop coating method. The resulting assay exhibited a good potentiometric response to different saccharides including glucose, and could be a promising application for the precise electrochemical detection of vicinal diols of specific sugars for clinical diagnostics, medicine validation, bioscience research and food analysis.
Introduction
Ultra specific saccharides sensors are highly desirable for accurate diagnosis of many diseases and monitoring of food quality.1,2 It is well known that glucose, fructose, lactose, saccharose, maltose, etc. are associated with several diseases, food quality and fermentation.3 For example, a high level of glucose in the blood – known as hyperglycemia – is a major risk factor for acute kidney failure, malfunction in nervous system, heart diseases, hypertension, damage to the retina, etc.4 In this essence, detection of saccharides is important for clinical diagnostics, medicine validation, bioscience research and food analysis. Among the reported methods, saccharides concentration is mainly determined using enzyme or chemical receptor techniques but they have a limited detection range mostly from 10−3–10−6 mol.5–10 Thus, there is a practical requirement to build up an ultra sensitive, selective, rapid and low-priced saccharides sensor having a detection limit in the nanomole range.The majority of existing saccharides sensors use monoboronic acid receptors, where the saccharide concentrations are estimated to the corresponding cyclic boronate ester yielded during the reaction using a fluorescent technique, however, this remains less sensitive, with many cases the limit of detection being at the 10−3 mol level.11,12 This is because of the low binding affinity of boronic acids with saccharides at 10−2–10−3 mol range of magnitude and/or the small difference in the reporting signals such as fluorescence quantum yield between the saccharide boronate ester and the boronic acid.13 It therefore shows that, in simple boronic acid based saccharide sensing, signal amplification corresponds to a basic and critical issue for improving sensitivity. We here report an ultra sensitive saccharides assay to achieve so by coupling the classic sensing strategy based on saccharides interaction to covalently attached boronic acid with acid functionalized chitosan (HOOC-chitosan) containing well-known Gd2O3:Eu3+ nanometre-sized rare earth phosphors particles, during the coupling reaction, boronic acid is a reactant. In this way, the original difference between the responsive physical signals of the corresponding yielded saccharides boronate ester is amplified by increasing the electron transfer kinetics of the medium.
On the other hand, chitosan is a relatively inexpensive and stable electroactive material that allows for the possible mass production of sensors. It is one of the most extensively used biopolymers in sensor applications due to its nontoxic behavior, excellent film forming ability, good mechanical strength, high permeability, and cost-effectiveness.14 Several enzymes have been successfully immobilized in chitosan/metal nanocomposite matrices and employed for sensing applications.15–18 In the case of developing ultra sensitive electrochemical sensors, introduction of metal nanoparticles is proven essential for the efficient electron transfer since direct electron transfer between sensing elements and electrode is not efficient in most cases because active sites are deeply embedded inside the massive sensing moieties and the controlled orientation of molecules on the electrode surface. Particularly, the physical properties of rare earths nanoparticlesviz. Gd, Eu, etc. are found quite attractive as well as sensitive to the structure and filling of the conduction bands due to their incomplete 4f shells, typically excess Gd leads to an antiferromagnet–ferromagnet transition adjunct with a semiconductor–metal transition.19 By this system, we could enhance the electronic surface response of electrodes. The rare earths nanoparticles can be physically embedded in or covalently attached to the matrix and significantly increase or tune the sensitivity of electrochemical sensors. Therefore, proper tuning of physical and chemical properties of the matrix is essential for maximizing the activity of the entrapped sensing moiety.
In the present study, we aimed to combine the merits of Gd2O3:Eu3+ nanophosphors and chitosan as a platform for ultra sensitive saccharides sensing using covalently amino phenyl boronic acid as a sensing element. The complete sensor systems retain their ultra fast activity due to facile mass transport property. In this way, we have synthesized Gd2O3:Eu3+ nanophosphor for making HOOC-chitosan nanocomposites. After that, the HOOC-chitosan/Gd2O3:Eu3+ nanocomposite matrix was used for covalent immobilization of amino phenyl boronic acid (APBA) and the resulting HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode could be offered a stable electrochemical saccharides sensory system in the 10−6–10−9 mol concentration(s) range.
Experimental
Materials
Chitosan (Mv = 1 × 106 g mol−1, DDA = 80–90%) was procured from Dainichiseika Color & Chemicals Mfg. Co. Ltd., Japan and used after purification. The following materials were used without further purification: Gd(NO3)3·6H2O (Aldrich, USA, 99.9%), Eu(NO3)3·6H2O (Aldrich, USA, 99.9%), oxalic acid dehydrate (E-Merck, Germany, 99%), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (WSC, Japan, 99%), N-hydroxysuccinimide (TCI, Japan, 99%), potassium ferrocyanide (Aldrich, USA, 99.9%), 3-aminophenylboronic acid (APBA, Alfa aesar, USA, 98%), D-glucose (TCI, Japan, 99%), D-galactose (TCI, Japan, 99%), D-fructose (TCI, Japan, 99%), D-fucose (TCI, Japan, 99%), D-lactose monohydrate (TCI, Japan, 99%), D-maltose monohydrate (TCI, Japan, 99%), D-saccharose (TCI, Japan, 99%), D-mannose (TCI, Japan, 99%), uric acid (TCI, Japan, 99%), urea (TCI, Japan, 99%), L-ascorbic acid (Sigma, USA, 95%), DL-malic acid (SAJ, Japan, 99%), L-alanine (Kabu, Japan, 99%), L-proline (Wako, Japan, 99%), sodium pyruvate (Sigma, USA, ≥99%) and sodium chloride (Wako, Japan, 99%, NaCl). All supplementary chemicals were of analytical grade and solutions were prepared with Milli-Q water. To prepare the chitosan solution, 0.8 g of chitosan flakes was dissolved into 100 mL of 90% acetic acid. The resulting mixture was stirred for 48 h at room temperature until the chitosan flakes were completely dissolved. The chitosan solution was stored in a refrigerator at 4 °C.Synthesis of Gd2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :Eu3+ nanoparticles
:Eu3+ nanoparticles
The stock solutions of metal nitrates (10 mM), i.e., Gd(NO3)3·6H2O and Eu(NO3)3·6H2O were prepared by dissolving appropriate amounts of metal nitrates (Eu3+ for 5% doping) in Milli-Q water. Simultaneously, 10 mM solution of oxalic acid was prepared separately. Then, 50 mL of Gd(NO3)3·6H2O stock solution and 50 mL Eu(NO3)3·6H2O stock solution were allowed to mix together homogeneously for 45 min. After that 50 mL of oxalic acid solution was added to the prepared metal nitrate mixture. After 30 min of stirring at room temperature, the solution was heated at 100 °C with constant stirring until a visible precipitate appeared. The precipitate was centrifuged, washed several times with absolute ethanol and Milli-Q water. Obtained precipitated slurry was kept in air for 24 h to get nanopowder of Gd2O3:Eu3+.
Synthesis of HOOC-chitosan
Carboxyl functionalized chitosan (HOOC-chitosan) was prepared via microwave assisted copolymerization followed by a hydrolysis technique.20 First, the chitosan was secured with graft-copolymerization of polyacrylonitrile onto chitosan. Next, chitosan-co-poly(acrylonitrile) was hydrolyzed in alkali medium in order to convert –CN into –COOH and –CONH2.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1454 (δC–H, -CH2-), 1362 (δCO, C–H), 1260 (δC–N), 1077 (δC–O) and 1032 (δO–H).
O), 1708 (νCO), 1456 (δC–H, -CH2-), 1361 (δCO, C–H), 1070 (δC–O) and 1034 (δO–H).
O), 1454 (δC–H, -CH2-), 1362 (δCO, C–H), 1260 (δC–N), 1077 (δC–O) and 1032 (δO–H).
O), 1708 (νCO), 1456 (δC–H, -CH2-), 1361 (δCO, C–H), 1070 (δC–O) and 1034 (δO–H).
Saccharides detection assay
The overall fabrication and detection routes are shown in Fig. 1. The saccharides detection assay was fabricated using a two steps process as described below:![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :Eu3+ nanocomposite electrode.
Ten mL of 1% (w/v) HOOC-chitosan solution was diluted with 4.5 mL of 0.25% acetic acid, and then 500 μL of 0.65% Gd2O3:Eu3+ nanoparticles solution was added. Next, the solution mixture was mechanically stirred (i.e., at 250 rpm) for 72 h at room temperature. After that, five μL of the resulting HOOC-chitosan/Gd2O3:Eu3+ nanocomposite solution was uniformly spread on the Pt-disc electrode by drop casting technique and dried at room temperature. The HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode was washed with Milli-Q water followed by a phosphate buffer saline (PBS) solution of pH 7.0 to neutralize the electrode surface.
:Eu3+ Pt-electrode surface using EDC/NHS as coupling reagent. To facilitate APBA covalent immobilization, the HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode was immersed in PBS (1M, pH 7.0) containing formerly activated APBA (100 mM) with 0.03 M 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and 0.03 M NHS for 2 h. The APBA immobilized electrode was rinsed with PBS at pH 7.0 in order to remove the excess unbound APBA. All experiments were carried out at room temperature. The APBA immobilized electrode was stored under dry conditions at 4 °C in a refrigerator.
:Eu3+ nanocomposite electrode.
Ten mL of 1% (w/v) HOOC-chitosan solution was diluted with 4.5 mL of 0.25% acetic acid, and then 500 μL of 0.65% Gd2O3:Eu3+ nanoparticles solution was added. Next, the solution mixture was mechanically stirred (i.e., at 250 rpm) for 72 h at room temperature. After that, five μL of the resulting HOOC-chitosan/Gd2O3:Eu3+ nanocomposite solution was uniformly spread on the Pt-disc electrode by drop casting technique and dried at room temperature. The HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode was washed with Milli-Q water followed by a phosphate buffer saline (PBS) solution of pH 7.0 to neutralize the electrode surface.
:Eu3+ Pt-electrode surface using EDC/NHS as coupling reagent. To facilitate APBA covalent immobilization, the HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode was immersed in PBS (1M, pH 7.0) containing formerly activated APBA (100 mM) with 0.03 M 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and 0.03 M NHS for 2 h. The APBA immobilized electrode was rinsed with PBS at pH 7.0 in order to remove the excess unbound APBA. All experiments were carried out at room temperature. The APBA immobilized electrode was stored under dry conditions at 4 °C in a refrigerator.
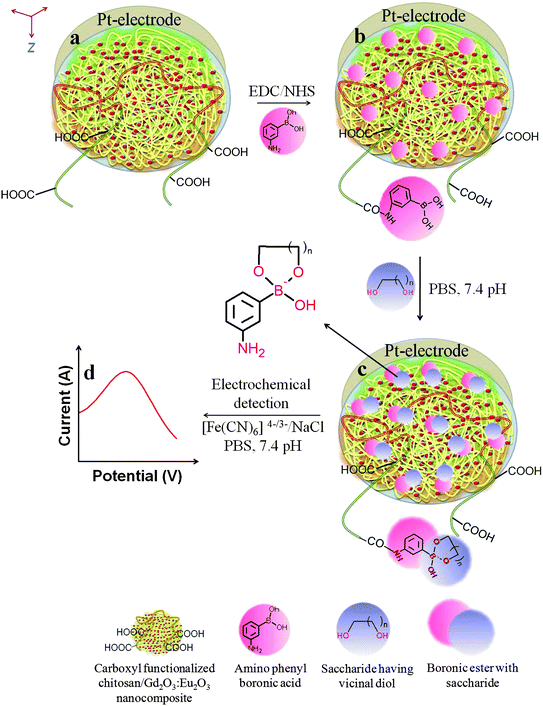 | ||
| Fig. 1 Schematic illustration of electrodes fabrication and electrochemical saccharide(s) detection process. | ||
Apparatus and instrumentation
The nanoparticles of Gd2O3:Eu3+ and HOOC-chitosan/Gd2O3:Eu3+ nanocomposite were systematically characterized using X-ray diffraction (XRD), energy dispersive X-rays (EDX) and transmission electron microscopy (TEM) to elaborate structural confirmation. XRD was performed on a Rigaku D/max-2200 PC diffractometer operated at 40 kV/40 mA, using CuKα1 radiation with a wavelength of 1.54 Å in the wide angle region from 20° to 60° on 2θ scale. The size and morphology of prepared nanoparticles were determined using a transmission electron microscope, model Tecnai 30 G2S-Twin electron microscope, operated at 300 KV accelerating voltage by dissolving the as-synthesized powder sample in ethanol and then placing a drop of this dilute ethanolic solution on the surface of a carbon coated copper grid.
A Milestone, PN44072, 1600 W laboratory microwave oven was used for graft copolymerization and hydrolysis reactions. FT-IR spectra were recorded on a Perkin Elmer SPECTRUM GX-Raman spectrophotometer. The surface morphology of the HOOC-chitosan/Gd2O3:Eu3+ and APBA containing HOOC-chitosan/Gd2O3:Eu3+ was examined with a Hitachi S-4800 field emission scanning electron microscope (SEM) operated at 5 kV. The specimens were sputter-coated with a thin layer of iridium (∼5 nm) prior to examination. The relative surface hydrophilicity of the HOOC-chitosan/Gd2O3:Eu3+ and APBA containing HOOC-chitosan/Gd2O3:Eu3+ was measured by automatic research grade contact angle measurement system, Kyowa interface science CA-W using water as liquid phase.
The electrochemical measurements were accomplished with an ALS/HCH 852CB electrochemical analyzer. A three electrode system was secured with a modified platinum disc electrode (Pt-electrode, OD: 6 mm, ID: 3 mm and PTE: 6 × 3 mm2), platinum wire, and Ag/AgCl (saturated KCl) as a working, counter and reference electrodes, respectively. A 50 mM PBS solution of pH 7.4 containing 2 mM Fe(CN)63−/4− and 0.25 mM was used as a redox electrolytic mediator. All measurements were carried out at 20 ± 2 °C.
Results and discussion
Synthesis and characterization of Gd2O3:Eu3+ nanoparticles, HOOC-chitosan and HOOC-chitosan/Gd2O3:Eu3+ nanocomposite
Fig. 2 (I) shows the XRD pattern of the Gd2O3:Eu3+ nanoparticles synthesized by the co-precipitation method. XRD spectra showed broad peaks at the positions of 26.15°, 31.94°, 35.14°, 40.46°, 42.03°, 47.97°, 52.50°, 53.60° and 59.07°, which were in excellent agreement with the standard JCPDS file for Gd2O3 (JCPDS 76-0155, a = b = c = 10.79 Å) and indexed as the body centered cubic structure of Gd2O3 having space group 1213 (199). All available reflections of the present phases were fitted with the Gaussian distribution. The broadening of XRD peaks (i.e. Scherrer's broadening) gave clear indication of nanosized formation of Gd2O3 and calculated average particle size using Scherrer's equation, i.e., <5 nm. Furthermore, in addition to the Gd2O3 phase, a new phase at 45.78° is also observed in the XRD spectra. This new phase in the XRD spectra corresponds to body centered cubic phase of Eu2O3 (521), i.e., matched with JCPDS 74-1988, which may be due to the formation of Eu2O3 from remaining unreacted Eu3+ ions present in the reaction solution.
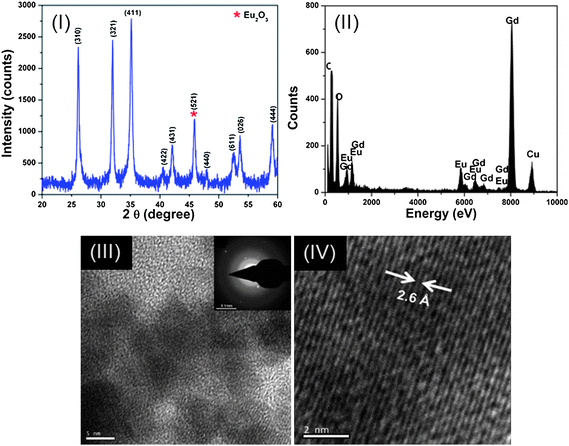 | ||
| Fig. 2 (I) XRD spectrum, (II) EDX spectrum, (III) and (IV) HR-TEM image of Gd2O3:Eu3+ nanoparticles. | ||
EDX measurements were further performed to confirm the doping and the involved valance states of gadolinium and europium in the synthesized Gd2O3:Eu3+ nanoparticles as shown in Fig. 2(II). From the similarity of the Gd and Eu peak intensity line traces, it is clear that after the synthesis process, gadolinium and europium are homogeneously distributed inside the nanoparticle. From the EDX line traces it can also be concluded that Eu3+ was successfully substituted into the crystal structure of Gd2O3. The estimated amount of Eu3+ ions was ∼4.7%. EDX measurements on single nanoparticle found that gadolinium and europium are homogeneously distributed throughout the Gd2O3:Eu3+ nanoparticle.
The morphology of the Gd2O3:Eu3+ nanoparticles was found to be nearly spherical in nature having diameters ∼4 nm. Fig. 2 (III and IV) clearly show that the diameters of these spherical nanoparticles were in agreement with those obtained using XRD results. Fig. 2 (III) shows the selected area electron diffraction (SAED) pattern. Broad diffused rings along with few diffraction spots are observed in SAED, indicating a mixture of amorphous and polycrystalline nature, which is obvious due to the nano-sized (∼4 nm) nature of the prepared sample. Fig. 2 (IV) show high-resolution transmission electron microscopy (HR-TEM) image of the Gd2O3:Eu3+ nanoparticles. The imaged lattice spacing 2.6 Å corresponds to the (411) plane of the cubic phase of Gd2O3, with a very slight deviation in the imaged lattice spacing obtained from XRD spectra. This deviation in imaged d-spacing corresponds to the induced lattice strain due to the formation of Eu2O3 phase from remaining unreacted Eu3+ ions in the reaction solution. This is in good agreement with already discussed XRD results.
Further, chitosan was grafted with acrylonitrile using microwave irradiation technique.22 Under the influence of microwave frequency, chitosan was randomly grafted with polyacrylonitrilevia free radical graft copolymerization mechanism. This can be attributed to the fact that the presence of a large number of pendent polar –OH and –NH2groups in chitosan may generate a fast microwave dielectric heating, which could rapidly provide macro and monomer free radicals for graft copolymerization reaction. Further, a sequence of HOOC-chitosan was yielded using base hydrolysis of prepared chitosan-co-poly(acrylonitrile) by means of conversion of –CN groups into –COOH and –CONH2groups. The chitosan-co-poly(acrylonitrile) showed characteristic peaks in FT-IR region: 1) –CN absorption at 2243 cm−1; 2) 2926, 2831 and 2717 cm−1 due to C–H stretching bands at –CH2–CN–, –CH2–, and –CH–, respectively; and 3) –CH2 deformation vibration at 1454 cm−1 confirmed the grafting of poly(acrylonitrile) onto chitosan, whereas after alkali hydrolysis of chitosan-co-poly(acrylonitrile), the peak at 2243 cm−1 had almost disappeared but an additional peak had arrived at 1708 cm−1 due to the C–O stretching of the –COOH group with a broadening at the amide band. The strong peak around 3400 cm−1 could be assigned to the stretching vibration of O–H of acid groups, the extension vibration of N–H, and inter-hydrogen bonds of the HOOC-chitosan.
HOOC-chitosan/Gd2O3:Eu3+ nanocomposite was fabricated by mechanical agitation of Gd2O3:Eu3+ nanoparticles into HOOC-chitosan solution. TEM, HR-TEM and SAED pattern of HOOC-chitosan/Gd2O3:Eu3+ nanocomposite is shown in Fig. 3. One can clearly observe the Gd2O3:Eu3+ nanoparticles in the HOOC-chitosan matrix. Whereas, HR-TEM image further confirms the formation HOOC-chitosan/Gd2O3:Eu3+ nanocomposite, although we are unable to see/resolve the lattice fringes. This could be attributed to the fact that these nanoparticles are coated by the HOOC-chitosan polymer and thus is not much crystalline in nature. This observation is also well supported by the corresponding SAED pattern. The SAED pattern shows broad-diffused rings, i.e., corresponding to the HOOC-chitosan polymer with few spots, i.e., corresponding to the Gd2O3:Eu3+ nanoparticle. Thus, this microscopic study effectively demonstrates the successful preparation of the HOOC-chitosan/Gd2O3:Eu3+ nanocomposite.
 | ||
| Fig. 3 (a) TEM image, (b) HR-TEM and (c) SAED pattern of HOOC-chitosan/Gd2O3:Eu3+ nanocomposite. | ||
Electrode fabrication and characterization
The prepared HOOC-chitosan/Gd2O3:Eu3+ nanocomposite was deposited upon Pt-disc electrode by dip-coating technique to form a uniform film (thickness <5 μm). The available carboxylic groups in the HOOC-chitosan/Gd2O3:Eu3+ were used for covalent immobilization of APBA using EDC and NHS as the coupling agents during the electrode preparation process.23 In this procedure, the amide bonds were formed between the amine groups of APBA and the carboxylic groups of HOOC-chitosan in the HOOC-chitosan/Gd2O3:Eu3+ matrix. Fig. 1 shows the overall steps for fabricating the APBA/HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode.
The ensuing APBA immobilized electrode was characterized by FT-IR spectroscopy. Fig. 4A shows the FT-IR spectra of the HOOC-chitosan/Gd2O3:Eu3+ and APBA/HOOC-chitosan/Gd2O3:Eu3+. The FT-IR spectrum of the HOOC-chitosan/Gd2O3:Eu3+ electrode (Fig. 4A, I) showed the characteristic peaks at: 1) about 3050 to 3600 cm−1 (O–H and N–H stretching); 2) 2953 and 2830 cm−1 (C–H stretching of –CH2groups); 3) 1728 cm−1 (CO stretching of carboxylic group); and 4) 1631 cm−1 (CO stretching of amide group). The CO characteristic peaks at 1728 and 1631 cm−1 confirm the presence of –COOH and –CONH2 in the HOOC-chitosan/Gd2O3:Eu3+ matrix. In addition, the absorption band at 3050 to 3600 cm−1 indicates the presence of substantial carboxylic groups on the HOOC-chitosan that are used to covalently immobilize the APBA.
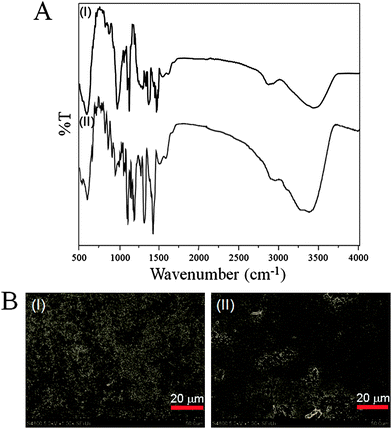 | ||
| Fig. 4 (A) FT-IR spectra of (I) HOOC-chitosan/Gd2O3:Eu3+ nanocomposite and (II) covalently attached APBA onto HOOC-chitosan/Gd2O3:Eu3+ nanocomposite; and (B) SEM image of (I) HOOC-chitosan/Gd2O3:Eu3+ nanocomposite and (II) covalently attached APBA onto HOOC-chitosan/Gd2O3:Eu3+ nanocomposite. | ||
The FT-IR spectrum of the APBA/HOOC-chitosan/Gd2O3:Eu3+ (Fig. 4A, II) showed peaks broadening at 1) 3147 to 3540 cm−1 (O–H and N–H stretching vibration); 2) 3051 cm−1 (C–H stretching of –CH2groups of benzene ring); 3) 2942 to 2856 cm−1 (C–H stretching of –CH2groups); 4) 1723 cm−1 (CO stretching of free carboxylic group); and 5) 1610 cm−1 (CO stretching of amide group) due to the attachment of APBA to HOOC-chitosan through the amide linkage in the HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode. Hence, FT-IR spectra confirm the covalent immobilization of APBA onto the HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode.
A typical SEM picture of HOOC-chitosan/Gd2O3:Eu3+ (Fig. 4B, I) exhibited minute Gd2O3:Eu3+ nanoparticle with a porous surface morphology. The porous surface of the HOOC-chitosan/Gd2O3:Eu3+ electrode provided a very high surface-to-volume ratio, which can provide the three-dimensional accommodation of APBA within HOOC-chitosan/Gd2O3:Eu3+ film of the electrode. Consequently, this will lead to a much higher level of APBA stability and much better APBA reproducibility for the APBA/HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode. Immobilization of the APBA over the HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode surface produced a homogeneous granular dendritic surface morphology (Fig. 4B, II). The uniform dendrite-like APBA electrode surface may be formed due to the covalent binding of APBA molecules over the Gd2O3:Eu3+ nanoparticle containing multi-functionalized chitosan, i.e., the HOOC-chitosan. To study the relative surface hydrophilicity, the contact angles of HOOC-chitosan/Gd2O3:Eu3+ and APBA/HOOC-chitosan/Gd2O3:Eu3+ electrodes were measured about 105° and 86.9°, respectively.24 The work of adhesion between the surface of the electrodes and the water droplet (Wa) can be calculated using the Young–Dupre eqn (1):
| Wa = γ(1 + cos θ) | (1) |
:Eu3+ Pt-electrode, this indicates that APBA/HOOC-chitosan/Gd2O3:Eu3+ electrode surface has a sturdier affinity to the hydrophilic moiety than HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode surface.
Electrochemical measurements
Fig. 5A shows the CVs of both HOOC-chitosan/Gd2O3:Eu3+ and APBA/HOOC-chitosan/Gd2O3:Eu3+at a constant 0.006 Vs−1 scan rate in PBS (pH 7.4, 0.25 mM NaCl, 2 mM Fe(CN)63−/4−). The CV of the HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode showed a decrease in current from 8.28 to 6.93 μA after the immobilization of APBA due to a slow redox process during the electrochemical reaction. Though, the current response of native HOOC-chitosan and APBA immobilized HOOC-chitosan Pt-electrodes were found in order of nA, likewise, the ratio of HOOC-chitosan Pt-electrode after the immobilization of APBA showed more or less identical current response. The result supports the claim that Gd2O3:Eu3+ nanoparticles provide a medium for fast electron transfer within the APBA/HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode. The oxidation (Ipa) and reduction (Ipc) peak current of both types of electrodes was also plotted against the √ν (where as ν is the scan rate), as shown in the inset.
 | ||
| Fig. 5 (A) Cyclic voltammograms of the (I) HOOC-chitosan/Gd2O3:Eu3+ and (II) APBA-HOOC-chitosan/Gd2O3:Eu3+ electrodes in PBS (pH 7.4, 2.5 mM NaCl, 2 mM Fe(CN)63−/4−) at different scan rates ranging from 0.001 to 0.012 Vs−1. Insets show oxidation and reduction peak currents with the square root of the scan rate. (B) A comparative plot of ΔE against the square root of the scan rate for electrode I and II. | ||
The peak current, i.e., Ipa or Ipc is proportional to ∛n√Dν at constant surface area of the HOOC-chitosan/Gd2O3:Eu3+ and APBA/HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode and saccharide(s) or analyte's concentration; n and D are the number of electrons appearing in the half-reaction for the redox couple and the analyte's diffusion coefficient, respectively. The peak current showed a linear response with the scan rate that is representing a diffusion controlled process, where the slope of the peak current with the square root of scan rate [d(I)/d√ν ∝ √D] depends on the diffusion coefficient. After immobilizing the APBA, the slope for the HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode increased by a factor of 1.21 compared with that of the bare HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode. This might be due to the covalent binding of APBA with the HOOC-chitosan/Gd2O3:Eu3+ electrode that encourage the moment of the supporting electrolyte's ions.25 The increase in the value of the current may be caused by the immobilization of ionic-APBA molecules onto the HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode surface.
Fig. 5B shows the CV in PBS (pH 7.4, 0.25 mM NaCl, 2 mM Fe(CN)63−/4−) for both electrodes at five different scan rates ranging from 0.001 Vs−1 to 0.012 Vs−1. The difference between the cathodic (Epc) and anodic (Epa) potential peak shift (ΔEp = Epa − Epc) was calculated and all ΔEp values were plotted against the square root of scan rates. Due to the slow kinetics of electron transfer on the electrode surface, a linear increase in ΔEp with an increase in the scan rate was observed.26 Immobilization of APBA on the HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode showed comparatively a lower electron kinetics response, thereby indicating a decrease in the number of electrons (n ∝ 1/ΔEp) involved in the reaction. In addition, an increase in the slope of ΔEp with the scan rate indicates that APBA may have captured some of the electrons involved in the reaction at higher scan rates because of slow electron transfer kinetics.
The surface concentration of covalently attached ABBA upon HOOC-chitosan/Gd2O3:Eu3+ electrode has been estimated by the Brown–Anson equation, i.e., ipa = n2F2I*AV/4RT, where, ipa is the peak current density in A cm−2; n is the number of electrons transferred; F is the Faraday constant (96584 C mol−1); I* is the surface concentration (mol cm−2) obtained for the ABBA/HOOC-chitosan/Gd2O3:Eu3+ nanocomposite matrix; A is the surface area of the electrode (1.8 cm2); V is the scan rate (0.006 Vs−1); R is the gas constant (8.314 J mol−1 K); and T is the absolute temperature (298 K). The value of surface charge concentration was calculated in the order of 6.93 × 10−3 mol cm−2. It is well known that I* usually depends on the electrode material as well as the immobilization process of the sensing element.27 The above results sustained that I* of the enzyme-free saccharides sensor has good capability to covalently accommodate APBA onto HOOC-chitosan/Gd2O3:Eu3+ nanocomposite matrix.
The impact of different saccharides complexation with aromatic boronic acids is illustrated by the significant increase in the ΔE values ranging from 0.512 to 9.21 mV in slight basic solution, i.e., at pH 7.4 upon the addition of eight saccharides (cf., D-glucose, D-galactose, D-fructose, D-fucose, D-lactose monohydrate, D-maltose monohydrate, D-saccharose and D-mannose). The increase in ΔE is a result of the formation of the respective saccharide boronate anion.28Fig. 6A and B demonstrate that the different saccharide boronate anion species is electrochemically active within the potential window between −0.3 and +1.2 V. In contrast, APBA is readily oxidized in the presence of electrolytes at approximately +1.0 V. The behavior of potential in the presence of saccharides and APBA results in the formation of corresponding boronate esters of different pKa values which grow on the electrode surface in concurrence with the previous report.29 The HOOC-chitosan/Gd2O3:Eu3+ nanocomposite immobilized ABBA (cf., Fig. 1) consists of a large numbers of gluco- amine groups. The distribution of these groups is in part a function of the degree of protonation of the matrix i.e. a function of the pH (pKa about 3.5 for protonated amine). Since the redox chemistry of HOOC-chitosan/Gd2O3:Eu3+ immobilized ABBA in the presence of saccharides involves electron and proton-transfer processes, the electrochemical potential is sensitive to changes in pH. More precisely, the dependence of the electrochemical potential on pH can be described by a double-square scheme. In this condition, the system appears to undergo a thermodynamically reversible 2e− ↔ 2H+ redox reaction, the Nernstian expression, which is a function of proton concentration and Ka:
| E = E°′NH3+/NH2 + (RT/2F) In[NH2][H+]2/Ka[NH2] | (2) |
 | ||
| Fig. 6 (A) Saccharide samples 1 to 8, i.e., 1) D-fructose, 2) D-fucose, 3) D-glucose, 4) D-galactose, 5) D-mannose, 6) D-lactose monohydrate, 7) D-saccharose and 8) D-maltose monohydrate response graph; and (B) saccharides concentration as a function of ΔE, i.e., obtained from the CV studies using a APBA-HOOC-chitosan/Gd2O3:Eu3+ electrode in PBS (pH 7.4, 2.5 mM NaCl, 2 mM Fe(CN)63−/4−) at a fixed scan rate of 0.006 Vs−1. | ||
Moreover, the influence of electrochemical potential of a proton coupled redox reaction, i.e., changes in the properties of a substituent group can be explained in two ways: (1) altering the formal potential of the redox couple, i.e., E°′NH3+/NH2 and (2) altering the acid dissociation constant, Ka. In the case of the conversion of the boronic acid to the boronate ester by cis-diol (Fig. 1), it is expected that there will be changes in both the inductive and resonance properties of the boron moiety. Specifically, formation of the boronate ester leads to an increase in its inductive electron donating ability, while eliminating the mesometrically electron withdrawing nature associated with the vacant p-orbital on the boron.30 Under conditions where, dE/dpH = 0. It means both oxidized and reduced forms are completely dissociated and as a result the increase in the electron-donating ability of a substituent will decrease the formal potential. This behavior has in fact been observed for boronic acid functionalized HOOC-chitosan/Gd2O3:Eu3+. On the other hand, increasing the electron donating ability of a substituent is expected to stabilize the acid form (i.e., NH3+). As such, converting boronic acid into the boronate anion esters are also expected to reduce the formal potential as well as to reduce the Ka of the protonated chitosan groups. It becomes apparent that the two processes offset one another as indicated in eqn (2), although the net effect will depend on the relative magnitude of the two influences and is ultimately expected to be a function of the binding constant and concentration of the analyte. The influence of both the transient pH change and the net thermodynamic effect of complexation on ΔE is shown in Fig. 6B where saccharides with varying binding constants (D-fructose >D-fucose >D-glucose >D-galactose >D-mannose >D-lactose monohydrate >D-saccharose and >D-maltose monohydrate) are added. The ΔE points generated upon addition of a saccharide are attributed to transient pH changes that occur upon complexation (cf., Fig. 1 and eqn (2)). The rapid decrease in the ΔE is likely due to diffusion of protons from the APBA/HOOC-chitosan/Gd2O3:Eu3+ nanocomposite Pt-electrode into the electrochemical cell solution. Therefore, it indicates that covalently attached ABBA containing HOOC-chitosan/Gd2O3:Eu3+ can provide a very high electroactive surface area for fast saccharides sensing with the lowest detection limit in the nanomole concentration range, i.e., 25 nM. A linear regression curve was observed for non-enzyme saccharides sensor responses for all selected saccharides concentration ranging from 25 nM to 13.5 μM onto APBA containing HOOC-chitosan/Gd2O3:Eu3+ nanocomposite Pt-electrode. A potentiometric linear response was observed with the successive addition of eight chosen saccharides in the PBS containing 0.25 mM NaCl as an electrolyte, and 2 mM Fe(CN)63−/4− as the redox moiety under a constant stirring at three to five-minute intervals. The electrode responded within three seconds with successive addition of different saccharides concentrations. The lowest detection limit of the HOOC-chitosan/Gd2O3:Eu3+ containing ABBA electrode towards saccharides concentrations was 25 nM. This new type of non-enzyme saccharides sensor demonstrated a shorter response time and a broader diluted detection range with respect to those reported previously using boronic acid as the saccharides sensing element as shown in Table 1. The small detection limit indicates a high affinity of APBA to the saccharides over the HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode surface, which may be attributed to 1) the advantageous nanoporous surface of the HOOC-chitosan/Gd2O3:Eu3+ nanocomposite electrode for the APBA immobilization that can favor quick conformational changes of the saccharides in electrochemical cell solutions, and 2) the high surface-to-volume ratio, which can help to effectively immobilize APBA onto the HOOC-chitosan/Gd2O3:Eu3+ nanocomposite Pt-electrode. Further, Gd2O3:Eu3+ rare earth nanoparticles could be provided a medium for efficient electron transfer between the active site of the APBA and the electrode, thereby enhancing the saccharides sensing activity in the 10−6–10−9 mol concentration range.
:Eu3+ saccharides sensor characteristics with previously reported boronic acid based saccharides sensors
| Probe materials | Immobilization method | Detection technique | Linear range | Detection limit | Response time | Reference |
|---|---|---|---|---|---|---|
| APBA/HOOC-chitosan/Gd2O3:Eu3+ nanocomposite |
Covalent | Potentiometric | 25 nM–13.5 μM | 25 nM | 3 s | Present work |
| PSA/PSSA electrospun fibers-mat | Physical adsorption | Amperometric | 0.75–14 mM | 0.75 mM | 4 s | [8] |
| Poly(acrylamidophenylboronic acid)-co-poly(methyl aminoethyl acrylate)/polystyrene sulfonate polyelectrolyte | — | UV-vis | 13.89–27.78 mM | 13.89 mM | 250 s | [31] |
| Au surface | Physical adsorption | SPR | 1 × 10−9−0.1 mM | 1 × 10−9 mM | 90 s | [32] |
| 9-[N-Methyl-N-(o-boronobenzyl)amino]methyl]anthracene and 9,10-bis[N-methyl-N-(o-boronobenzyl)amino]methyl]-anthracene | — | Fluorescence | 0.1–2.5 mM | 0.1 mM | — | [33] |
The reproducibility of the APBA/HOOC-chitosan/Gd2O3:Eu3+ nanocomposite Pt-electrode was investigated at a fixed condition, i.e., 350 nM saccharides concentration in the PBS containing 0.25 mM NaCl, and 2 mM Fe(CN)63−/4− under a constant stirring at three-minute intervals. After five uses, no significant drop on/off in current response was observed with the testing of same electrode; thus, the APBA electrode displayed good reproducibility. While, in a series of ten APBA electrodes, a relative standard deviation was found to be ±14.72%, i.e., obtained from the individual current response of the same sensor system. The good reproducibility observed with the saccharides sensor may be attributed to the efficient bonding of the APBA with the HOOC-chitosan/Gd2O3:Eu3+ functional probe. The interference effects of uric acid, L-ascorbic acid, sodium pyruvate, urea, DL-malic acid, L-alanine, L-proline and sodium chloride on the potentiometric response of APBA/HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode was demonstrated by adding the individual substance during the sensing experiment of their physical concentration, i.e., 75 nM. It was examined that interferents comprised an error of about ±8.34% by this technique (Table 2). Thus, HOOC-chitosan/Gd2O3:Eu3+ nanocomposite Pt-electrode can detect saccharides with the least interference.
:Eu3+ Pt-electrode saccharides sensor
| Analyte(s)/Interference | Potential response/mV cm−2 | Error (%) | |
|---|---|---|---|
| With interference | Without interference | ||
| 350 nM/dL glucose | — | 5.27 | 0.00 |
| 350 nM/dL glucose + 75 nM uric acid | 5.40 | 5.27 | 2.40 |
| 350 nM/dL glucose + 75 nM L-ascorbic acid | 5.59 | 5.27 | 6.07 |
| 350 nM/dL glucose + 75 nM sodium pyruvate | 5.71 | 5.27 | 8.34 |
| 350 nM/dL glucose + 75 nM urea | 5.38 | 5.27 | 2.09 |
| 350 nM/dL glucose + 75 nM DL-malic acid | 5.39 | 5.27 | 2.28 |
| 350 nM/dL glucose + 75 nM L-alanine | 5.28 | 5.27 | 0.19 |
| 350 nM/dL glucose + 75 nM L-proline | 5.31 | 5.27 | 0.76 |
| 350 nM/dL glucose + 75 nM sodium chloride | 5.35 | 5.27 | 1.52 |
| 350 nM/dL glucose in urine | 5.74 | 5.27 | 8.92 |
| 350 nM/dL glucose in blood serum | 5.79 | 5.27 | 9.88 |
To demonstrate the real-world application of the APBA/HOOC-chitosan/Gd2O3:Eu3+ nanocomposite Pt-electrode sensor for saccharides analysis, samples of fresh blood serum and urine from a healthy person were examined. The analysis was performed without any sample pre-treatment and results were compared with those obtained from the standard method.34 It was observed that the value of saccharide (e.g., glucose) concentration in blood serum and urine obtained with APBA/HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode sensor system was about 5–10% higher than those measured by the standard method, almost certainly because of interferences of electroactive species present in the biological samples (Table 2). On the whole, a good conformity of the saccharide concentration was observed in the both cases.
Conclusion
A new type of saccharides sensor based on a covalent immobilized APBA in thin films of HOOC-chitosan containing <5 nm Gd2O3:Eu3+ nanoparticles at a platinum disc electrode was successfully fabricated and assessed. The APBA/HOOC-chitosan/Gd2O3:Eu3+ Pt-electrode surface offered a high sensitivity to saccharides at nano to micro molar concentration range with a surface charge concentration of 6.93 × 10−3 mol cm−2. The saccharides sensor showed a linear potential response to the saccharides concentration ranging from 25 nM to 13.5 μM and it exhibited a sensitivity of 0.28 mV with a response time of three seconds. This new type of saccharides biosensor has demonstrated superior performance compared with those reported previously, including a higher sensitivity, wider range of detection limit in a nonomolar concentration range, and responded within a few seconds. With APBA as a model, the HOOC-chitosan/Gd2O3:Eu3+ nanocomposite-modified Pt-electrode could have potential for quick detection of saccharides from biological samples with minimum interference. Hence, the proposed sensor system has wide applications especially for use in medical and food technologies.
Acknowledgements
The authors offer their heartfelt gratitude to the National Institute for Materials Science, Japan for providing an infrastructure facility and to the JSPS, JST-CREST and MEXT Japan for their generous financial support to carry out this research.References
- D. A. Gough and J. C. Armour, Diabetes, 1995, 44, 1005 CAS.
- G. S. Wilson and Y. Hu, Chem. Rev., 2000, 100, 2693 CrossRef CAS.
- S. Vaidyanathan, G. Macaloney, J. Vaughan, B. McNeil and L. M. Harvey, Crit. Rev. Biotechnol., 1999, 19, 277 CAS.
- B. J. Hoogwerf, Int. J. Diabetes Dev. Countries, 2005, 25, 63 Search PubMed.
- T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed. Engl., 1996, 35, 1910 CrossRef.
- N. DiCesare, M. R. Pinto, K. S. Schanze and J. R. Lakowicz, Langmuir, 2002, 18, 7785 CrossRef CAS.
- J. Yoon and A. W. Czarnik, J. Am. Chem. Soc., 1992, 114, 5874 CrossRef CAS.
- A. Tiwari, D. Terada, C. Yoshikawa and H. Kobayashi, Talanta, 2010, 82, 1725 CrossRef CAS.
- X. Kang, J. Wang, H. Wu, I. A. Aksay, J. Liu and Y. Lin, Biosens. Bioelectron., 2009, 25, 901 CrossRef CAS.
- L. Luo, Q. Li, Y. Xu, Y. Ding, X. Wang, D. Deng and Y. Xu, Sens. Actuators, B, 2010, 145, 293 CrossRef.
- S.-Y. Xu, Y.-B. Ruan, X.-X. Luo, Y.-F. Gao, J.-S. Zhao, J.-S. Shen and Y.-B. Jiang, Chem. Commun., 2010, 46, 5864 RSC.
- D. K. Scrafton, J. E. Taylor, M. F. Mahon, J. S. Fossey and T. D. James, J. Org. Chem., 2008, 73, 2871 CrossRef CAS.
- A. Schiller, R. A. Wessling and B. Singaram, Angew. Chem., Int. Ed., 2007, 46, 6457 CrossRef CAS.
- V. K. Mourya, N. N. Inamdar and A. Tiwari, Adv. Mater. Lett., 2010, 1, 11 Search PubMed.
- S. Cao, R. Mishra, S. Pilla and S. Tripathi, et al, Carbohydr. Polym., 2010, 82, 189 CrossRef CAS.
- D. Feng, F. Wang and Z. Chen, Sens. Actuators, B, 2009, 138, 539 CrossRef.
- A. Kaushik, R. Khan, P. R. Solanki, P. Pandey, J. Alam, S. Ahmad and B. D. Malhotra, Biosens. Bioelectron., 2008, 24, 676 CrossRef.
- J.-D. Qiu, R.-P. Liang, R. Wang, L.-X. Fan, Y.-W. Chen and X.-H. Xia, Biosens. Bioelectron., 2009, 25, 852 CrossRef CAS.
- N. A. Frey, S. Peng, K. Cheng and S. Sun, Chem. Soc. Rev., 2009, 38, 2532 RSC.
- V. Singh, A. Tiwari, D. N. Tripathi and R. Sanghi, J. Appl. Polym. Sci., 2004, 92, 1569 CrossRef CAS.
- V. Singh, A. Tiwari, S. Pandey, S. K. Singh and R. Sanghi, J. Appl. Polym. Sci., 2007, 104, 536 CrossRef CAS.
- V. Singh, A. Tiwari, D. N. Tripathi and R. Sanghi, Carbohydr. Polym., 2004, 58, 1 CrossRef CAS.
- A. Tiwari, S. Aryal, S. Pilla and S. Gong, Talanta, 2009, 78, 1401 CrossRef CAS.
- A. Abbasian, S. R. Ghaffarian, N. Mohammadi and D. Fallahi, Colloids Surf., A, 2004, 236, 133 CrossRef CAS.
- A. M. Lazarin and C. Airoldi, Anal. Chim. Acta, 2004, 523, 89 CrossRef CAS.
- A. Tiwari and S. Gong, Electroanalysis, 2008, 20, 2119 CrossRef CAS.
- P. R. Solanki, S. K. Arya, S. P. Singh, M. K. Pandey and B. D. Malhotra, Sens. Actuators, B, 2007, 123, 829 CrossRef.
- A. Ori and S. Shinkai, J. Chem. Soc., Chem. Commun., 1995, 1771 RSC.
- E. Shoji and M. S. Freund, J. Am. Chem. Soc., 2002, 124, 12486 CrossRef CAS.
- D. S. Matteson, in Progress in Boron Chemistry, R. J. S. H. Brotherton, Ed., Pergamon Press, New York, 1970, Vol. 3; pp 117–176 Search PubMed.
- B. G. D. Geest, A. M. Jonas, J. Demeester and S. C. De-Smedt, Langmuir, 2006, 22, 5070 CrossRef.
- H. Chen, M. Lee, J. Lee, J.-H. Kim, Y.-S. Gal, Y.-H. Hwang, W. G. An and K. Koh, Sensors, 2007, 7, 1480 CrossRef CAS.
- N. DiCesare and J. R. Lakowicz, Anal. Biochem., 2001, 294, 154 CrossRef CAS.
- L. L. Salomon and J. E. Johnson, Anal. Chem., 1959, 31, 453 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |