Mycobacterium tuberculosis detection via rolling circle amplification†
Eric
Schopf‡
a,
Yang
Liu
a,
Jane C.
Deng
b,
Siyin
Yang
a,
Genhong
Cheng
b and
Yong
Chen
*a
aDepartment of Mechanical and Aerospace Engineering, California NanoSystems Institute, University of California, Los Angeles, California 90095, USA. E-mail: yongchen@seas.ucla.edu
bDepartment of Microbiology, Immunology and Molecular Genetics, University of California, Los Angeles, California 90095, USA
First published on 2nd December 2010
Abstract
Hybridization-based assays for DNA detection often use single-stranded DNA (ssDNA) probes to capture ssDNA targets in solution. Unfortunately, these assays are often not able to detect double-stranded DNA (dsDNA). Here, we achieve highly sensitive dsDNA target detection by including short oligonucleotide sequences during denaturing and cooling. After performing an isothermal nucleic acid amplification technique (Rolling Circle Amplification, RCA), these captured dsDNA targets are labeled, allowing single amplified molecules to be imaged and counted. This detection method was first applied to the detection of PCR-generated (polymerase chain reaction) dsDNA targets, yielding a limit of detection of 4.25 fM. As an application of the developed assay, the detection of extracted Mycobacterium tuberculosis (M. tb.) genomic DNA was attempted. A M. tb.-specific target was detected with high specificity compared to similar bacteria, and a detection limit of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 colony forming units (cfu) ml−1 was achieved, close to the sensitivity required for clinical diagnosis.
000 colony forming units (cfu) ml−1 was achieved, close to the sensitivity required for clinical diagnosis.
1. Introduction
While there is an unquestionable necessity for reliable, sensitive, and specific DNA detection assays, most published work focuses on the detection of single-stranded DNA (ssDNA) targets.1–6 These synthesized targets are simple to work with and straightforward to detect, as they are readily available for hybridization with other oligonucleotide probes. While useful in probing for things such as single nucleotide polymorphisms,7,8 these ssDNA assays have significant limitations when applied to double-stranded DNA (dsDNA) or genomic DNA targets, such as detecting DNA from clinical samples. These complex targets present a variety of challenges towards a hybridization detection assay. Paramount of these is the necessity to denature the target before its hybridization with capture DNA probes, while at the same time preventing the target re-hybridizing with its complementary strand. This is mainly achieved by means of microarray-based methods. However, many of these techniques involve amplification of the genomic DNA target by the polymerase chain reaction (PCR) before detection.9–13Non-microarray-based methods for detection of dsDNA targets have also been investigated. Fiber optic genomic DNA assays14 and site-specific nicking reactions leading to amplification15 have been demonstrated to detect genomic DNA targets. Other examples involve detecting dsDNA sequences directly through DNA-binding proteins, which has not yet shown the sensitivity needed for clinical diagnosis.16,17 Recently, a method to solve the re-hybridization of the dsDNA target was demonstrated by adding small “blocking oligos” to prevent the two strands of the dsDNA target from hybridizing together.18
Here, we present our developed assay for genomic dsDNA detection based on combining blocking oligonucleotide sequences with rolling circle amplification (RCA). RCA is an isothermal amplification technique in which a polymerase extends the length of a DNA molecule by several orders of magnitude using a circular primer as the template.4 These extended single target molecules tend to form dense clusters in solution on the order of a micron in size.19,20RCA is well studied, and a wide variety of oligonucleotide amplification and detection methods based on RCA have been reported.21–24 The function of the circular primers in our assay is two-fold. First, they can be added in excess and will rapidly hybridize with the targeted DNA. In this manner, they act as “blocking oligos” to sterically hinder the two strands of the dsDNA target from re-hybridizing. Along with capture probes, the excess of these sequences allows for these probes to effectively compete for hybridization with the dsDNA target against the complementary dsDNA strand. Second, circular primers can be used to extend the length of the targeted DNA strand by several orders of magnitude. As demonstrated in previous work, the elongated target DNA molecule can hybridize with many fluorescent oligonucleotide probes. Single target molecules can therefore be detected straightforwardly, as their fluorescence is many times brighter than the background.19,20
The inclusion of functional blocking oligonucleotide sequences to our previously described ssDNA detection assay25 demonstrates that the capture and detection of dsDNA target molecules, even those extracted from bacteria, can be achieved at very low concentrations without a pre-detection amplification step. The incorporation of RCA into the assay allows the sensitivity to exceed that of previously reported methods, thereby allowing individual amplified target molecules to be observed and femtomolar concentrations of dsDNA targets to be detected. Using this method, we show that the detection of extracted genomic DNA of the bacterial pathogen Mycobacterium tuberculosis (M. tb.) is possible without using PCR. Finally, the use of four DNA probes per dsDNA target allows for a high specificity for the designed target sequence, as confirmed through testing of other bacterial genomic DNA.
2. Experimental
2.1. Materials
SuperBlock T20 blocking buffer was purchased from Fisher Scientific. Amine reactive beads were extracted from GE Healthcare HiTrapTM NHS-activated HP 1 ml affinity column. RepliPhi(tm) Φ29 Polymerase was purchased from Epicentre Biotechnologies. T4 DNA Ligase (400000 U ml−1) and restriction enzymes PvuII and NaeI were purchased from New England BioLabs. DNA molecules were synthesized and purified at the Protein and Nucleic Acid Facility in the Stanford School of Medicine. If not specified, the rest of the chemicals used in the assay were purchased from Sigma Aldrich.
Blocking buffer was prepared by adding polyinosinic-polycytidylic acid potassium salt (dI-dC) to SuperBlock T20 blocking buffer at a final concentration of 50 μg/ml. Φ29 buffer was prepared in a higher concentration stock solution and then diluted to 1×: 50 mM Tris HCl, 10 mM MgCl2, 10 mM (NH4)2SO4, and 4 mM DTT, pH 7.5. 1× ligation buffer without ATP was prepared as 50 mM Tris HCl, 10 mM MgCl2, 10 mM DTT, pH 7.5. 1× PBS was prepared from packets from Sigma Aldrich and adjusted to either pH 7.4 or 8.0. DNA activation buffer was prepared as 20 mM potassium phosphate, dibasic, pH 8.0. Deactivation buffer was prepared as 0.5 M ethanolamine, 0.5 M NaCl, pH 8.3. Storage buffer was prepared as 50 mM Tris-HCl, 0.02% Tween-20, 0.02% sodium azide, pH 8.0.
Synthetic DNA sequences were dissolved in water upon arrival and stored at −20 °C. Serial dilutions were then prepared in water using previously blocked centrifuge tubes and stored at −20 °C with the exception of both fluorescent DNAs, which were added at 20 μM into 1× PBS pH 7.4 and stored without light at 4 °C. Table 1 lists the DNA sequences used in our assay. Note that each dsDNA target, made up of a forward and reverse strand, is able to hybridize with four different oligonucleotides: two capture probes and two RCA padlock primers. PCR primers were used to generate the 161 bp dsDNA target used in experiments.
| Name | Oligonucleotide sequence, 5′ to 3′ | ||
|---|---|---|---|
| Capture probes and RCA primers for PCR-amplified DNA targets | |||
| PCR capture 1 | CCTATCCGTATGGTGGATAAAAAAAAAAAAAAAAAAAAAAA-(NH2) | ||
| PCR capture 2 | GTCGGAGCGTCGGAAGCTCAAAAAAAAAAAAAAAAAAAAA-(NH2) | ||
| PCR RCA primer 1 | P-TGCATTGTCATAGGAGCGTTACTAGGTTACTGAGCAGACAACGAGGACACGCGTTACTTCGATCGATCGTCTCGGCTAG | ||
| PCR RCA primer 2 | P-GACCTGAAAGACGTTAGCTAGCTTAATCCTTTACGACGACACAGGACTACCTTCTAATTCGATCGACAAGAAGGCGTACTC | ||
| Capture probes and RCA primes for M. tb. genomic DNA targets | |||
| Genomic capture 1 | GATTCTTCGGTCGTGGTGGTCCCAAAAAAAAAAAAAAAAAAAA-(NH2) | ||
| Genomic capture 2 | GGTTCATCGCCGATCATCAGAAAAAAAAAAAAAAAAAAAAA-(NH2) | ||
| Genomic RCA primer 1 | P-GACTCGACACCCCACCAAATTCGATCATATTCTGAGCACAACGAGGATCAATCGTATCGAACAGCTGTGTGCAGATC | ||
| Genomic RCA primer 2 | P-GGACCACGACCGAAGATGTATCATATACTATACGACGACAGGACTACCATAACTAACCAAACCGCCGGCGCACGGCCCG | ||
| DNA fluorescent oligonucleotide labels | Primers for PCR target generation | ||
| Label 1 | (FAM)-GAGCAGACAACGGACACG | Forward primer | ACAAGAAGGTACTCGACCTGAA |
| Label 2 | (Cy3)-TACGACGACAGGCTACC | Reverse primer | TGATCGTCTCGGCTAGTGCATTGT |
2.2. Capture bead preparation
Sepharose beads (mean diameter: 34 μm) were extracted from an affinity column with a clean razor blade and immediately suspended in 1 ml of 100% ethanol. For bead activation, 100 μl of activation buffer was added to 200 μl tubes along with 10 μl of beads stored in ethanol. The beads were rinsed quickly . The rinsing procedure for all of the steps described in this protocol consisted of a gentle vortex to suspend the beads, followed by bench top centrifugation for 10 s. The tubes were then rotated by 180° and centrifuged again for 10 s. This was followed by removal of 100 μl of solution, leaving approximately 10 μl of solution at the bottom of the tube. After rinsing, 80 μl of activation buffer and 5 μl of 50 μM capture probes were added to each reaction tube. Following 1 h of incubation on a rotator to ensure bead suspension, the tubes were rinsed twice in deactivation buffer. After the addition of 90 μl of deactivation buffer the tubes were again incubated on a rotator for up to 2 h. The tubes were then rinsed twice in storage buffer, suspended in 100 μl of storage buffer, and stored in a refrigerator at 4 °C.2.3. DNA target capture
100 μl of blocking buffer was added to 200 μl tubes and subsequently, 6 μl of activated beads was added. This was rinsed once with blocking buffer and suspended in 100 μl of storage buffer; the tubes were then allowed to incubate overnight on a rotator. Before target addition, the tubes were rinsed twice in 1× Φ29 buffer and the beads were then suspended in 10 μl of 1× Φ29 buffer. 1 μl of each RCA padlock primer at 5 μM was added to this solution. To this mixture, different amounts of target molecules were added. For PCR-target detection, 1 μl of different dsDNA concentrations was added. For detection of samples prepared from various concentrations of bacteria as well as the specificity experiments for S. pneumoniae and M. intercellulare, 10 μl was added. For a further description of the specificity experiments, see the ESI.† 1× Φ29 buffer was used to achieve a final reaction volume of 100 μl.To denature the dsDNA targets, a simple heating procedure was followed. Tubes with functionalized beads, target molecules, and RCA padlock primers were heated to 95 °C for 5 min. Halfway through as well as at the end of the procedure, the tubes were vortexed to ensure bead suspension. The tubes were then cooled to 50 °C over approximately 90 s, after which they remained at 50 °C for two minutes and were vortexed halfway through this step. The tubes were then transferred to the rotator which was placed in a 50 °C oven to maintain a constant temperature. This was allowed to proceed for approximately 17 h on average.
2.4. DNA amplification and detection
To ligate the padlock probes after the capture step, the beads were rinsed three times in 1× ligation buffer without ATP. 9 μl of 1× ligation buffer with ATP (provided by manufacturer) and 1 μl of T4 DNA ligase were added to 10 μl of the beads and buffer solution. The tubes were vortexed and allowed to react on a rotator for 15 min. After two rinses with 1× Φ29 buffer, RCA reagents were added to the reaction: 3.2 μl of 5× Φ29 buffer, 1.6 μl of 1% BSA, 1.28 μl of 10 mM deoxynucleotide triphosphates (dNTPs), and 0.64 μl of Φ29 polymerase at 10 U μl−1. The tubes were then vortexed to suspend the beads and transferred to a 37 °C oven, where they were allowed to incubate for 3 h on a rotator.Following amplification, the tubes were rinsed twice with 1× PBS. Fresh fluorescent DNA probes were prepared by diluting a 20 μM stock 10-fold into a total volume of 200 μl , followed immediately by centrifugation of the DNA at 9000 rpm for 10 min. 3 μl each of FAM and Cy3 fluorescent DNA probes were then added to the 10 μl bead solution. The tubes were then placed on a rotator in the dark for approximately 18 h. Afterwards, the tubes were rinsed twice in storage buffer and the beads were suspended in a final volume of 16 μl storage buffer. To prepare samples for imaging, the tubes were vortexed to suspend the beads, and 8 μl of the bead solution was dropped onto a glass slide. A glass coverslip was then immediately placed on top, spreading out the beads. One slide of bead solution was then imaged at each concentration and subsequently analyzed.
A Photometrics CoolSnap HQ2 camera was used with a 10× objective to take all the images using NIS Elements Basic Research v3.0 software. For each image location, a bright field image was first taken to determine bead locations. Next, a Cy3 image was taken with the appropriate filter. Finally, a FAM image was taken with the appropriate filter. Ten image locations were taken for each concentration. The total number of beads present in each image varied slightly, with an average of about 45 beads. The total number of counted target molecules in each of the ten images at each concentration was determined. An overall number of counted molecules per bead was established in each image. The two highest and two lowest counted target molecules per bead values were eliminated. An average value was then generated based on the remaining six images, and plotted with standard deviation, for each concentration. A description of the counting software can be found in the ESI.†
3. Results and discussion
3.1. Double-stranded DNA detection
A schematic of our assay is shown in Scheme 1. Sepharose beads functionalized with oligonucleotide capture probes serve as a detection platform. Beads are added in the reaction tube concurrently with dsDNA targets and RCA padlock probes, and the tube is heated to 95 °C for 5 min to denature each dsDNA into two complementary single strands, which sequentially hybridize with their capture probes as well as padlock probes after the tube is rapidly cooled to 50 °C. Following capture, the padlock probes are ligated into circular primers for use in RCA. Target strands are then extended by RCA at 37 °C, producing a small cluster of DNA. Both RCA-extended strands have different sequences that are designed to hybridize with two distinct fluorescently-labeled (FAM and Cy3) oligonucleotide probes for imaging. Once labeled with the appropriate fluorescent probes, beads are sandwiched between a glass slide and cover slip and imaged with a fluorescent microscope. To assist with unbiased counting, a software program was developed to automatically recognize and count each labeled single target molecule on the surface of the sepharose beads. | ||
| Scheme 1 Schematic of the dsDNA detection assay. Sepharose beads are conjugated with DNA capture probes. Double-stranded DNA targets are denatured by heating, and then hybridized with their corresponding capture and padlock probes after cooling. Following a ligation step, target molecules are extended by RCA and labeled with fluorescent (FAM and Cy3) probes. The duration of each step is indicated in bold. | ||
The sensitivity of our developed assay for dsDNA targets was investigated using 161 bp PCR-generated targets. Reaction tubes with different serially-diluted dsDNA target concentrations were prepared, and the assay was performed as described previously. After the reaction, the beads were imaged three times in succession. First, a bright field image was taken to record the locations of the beads. Two fluorescent images were then taken, one for each respective fluorescent label (FAM and Cy3). A typical superposition of these three images is shown, along with an output from the counting program (Fig. 1a). Amplified single DNA target molecules, appearing as red or green “dots”, are clearly visible on the bead surface. At each target concentration, ten separate image locations were gathered and the total number of amplified target molecules was counted; this is shown as a function of target concentration (Fig. 1b). The number of amplified target molecules increases with target concentration until the surface of the beads becomes saturated with these “dots” (Fig. 1b, Inset). From the graph (Fig. 1b), it can be estimated that the limit of detection of dsDNA target molecules is approximately 4.25 fM, which corresponds to approximately 2550 targets per microliter in the assay.
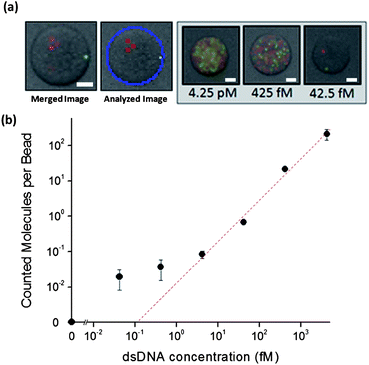 | ||
| Fig. 1 Detection of dsDNA targets. (a) A typical superposition of three images (bright field, FAM, Cy3) is shown. A software processed image of the same bead is shown in the middle. Superimposed images of beads at different target concentrations are shown on the right. (b) The counted amplified molecules per bead versus number of dsDNA targets shows a linear relationship (R2 = 0.996) above 4.25 fM. Error bars are ± s.d. (n = 6). Scale bar = 10 μm. | ||
It is important to note that each amplified target molecule for low target concentrations appears similar to those from higher concentrations in both size and intensity. This is due to the uniform amplification procedure, which creates a similar amplified cluster irrespective of the number of target molecules. However, at very high target molecule concentrations, the bead surface becomes saturated with amplified target molecules. Each bead would be entirely covered in these spots, hampering the software's ability to process the image, as the boundary between amplified target molecules is poorly-defined (data not shown). Similarly, at these high concentrations, steric hinderance between neighboring growing amplified target molecules slows their growth and prevents them from reaching a similar size to un-hindered target molecules. For these reasons, higher target concentrations were not investigated.
Unlike previously reported methods,14–17 dsDNA targets are captured by hybridization with very high sensitivity. The presence of the padlock and capture probes in the solution enables the dsDNA targets to be both captured and amplified. It is important to note that the capture of target strands only occurs at one time, during the incubation with capture probe-functionalized beads and padlock primers. Moreover, this step is done at an elevated, constant temperature (50 °C). This higher temperature serves two purposes. First, it increases the diffusion rate of targets and probes, which increases the capture rate onto the bead surface. Second, and more importantly, the higher temperature reduces unwanted secondary structures of the probes or targets from forming. These unwanted structures include hairpins and self dimers which can act to sterically hinder the capture reaction;12 by increasing the reaction temperature, these unwanted structures will not be formed, allowing for a more efficient capture reaction. Furthermore, the capture and padlock probes are not able to hybridize incorrectly with other parts of the target sequence, as 50 °C is above the melting temperature of these incorrect structures while simultaneously being significantly (>10 °C) lower than the correct hybridization melting temperature according to DINAMelt software.26 An elevated temperature for target incubation has been used on many occasions to enable hybridization between PCR-generated products and ssDNA capture probes.9,11–13 A limit of detection of 4.25 fM for dsDNA targets is a significant improvement upon reported sensitivities for dsDNA assays.
Finally, the entire assay is done as one linear process. No cycling or repeating of steps is necessary, simplifying the procedure and reducing the hands-on time required for the assay when compared to other techniques such as primer generation-rolling circle amplification.23 Furthermore, since only one amplification step is performed, any non-specific amplification is limited. In PCR, non-specific amplification can lead to a significant rate of false positive diagnosis27 as many amplification cycles are performed, each one with the potential to generate amplified products. However, further optimization of the assay would be necessary for it to compete with PCR in terms of speed and sensitivity.28
3.2. M. tb. DNA detection
As an application of this assay, the detection ofM. tb.genomic DNA was attempted. A M. tb.-specific region in the IS6110 element29 was selected for capture after cell lysis, DNA extraction, and purification. Extremely long and double stranded, M. tb.genomic DNA was fragmented by two restriction enzymes into small sections in order to be amplified.30 RCA must proceed from the 3′ end of a free DNA sequence, and enzymatic digestion allows the generation of free 3′ ends that can be subsequently amplified. Restriction enzymes NaeI and PvuII were chosen to generate a specific 227 base pair (bp) dsDNA target, from within the IS6110 element (Fig. 2a). As shown in Fig. 2b, a wide range of DNA fragments are generated by digesting genomic DNA (over four million bp) with both restriction enzymes. | ||
| Fig. 2 Preparation of M. tb.genomic DNA targets. (a) A schematic of the procedure for generating dsDNA targets from genomic DNA extracted from M. tb. bacteria. (b) Enzyme digestion of M. Tb.genomic DNA is shown on a 1% agarose gel. In lane 1 and 2, extracted and purified genomic DNA is shown. In lanes 3 and 4, the same genomic DNA after digestion with NaeI and PvuII restriction enzymes is shown. On the left, a ladder is shown with sizes for comparison. | ||
While the total DNA concentration can be measured, the exact concentration of the 227 bp target molecules is far less than the measured concentration, even when the multiple copies of the insertion element that can be found in the same bacterial genome are taken into account. It is important to note that at this point, all of these fragments could be used for detection, but only the specific 227 bp target for which the capture and padlock probe sequences were designed for will be detected by the assay. While the specific 227bp target was chosen to be contained within the IS6110M. tb.-specific region, the sequences of the padlock primers and capture probes were chosen based on various criteria mentioned in section 3.1: strong hybridization energies with the target sequence so as not to be denatured during the elevated incubation temperature step, distinct sequences to eliminate the possibility of incorrect hybridizations, and chosen so that any self-dimer or hairpin structures are unfavorable.
Cultures of M. tb. bacteria were prepared as described in the Methods section at concentrations ranging from 2.5 × 106 cfu ml−1 to 10 cfu ml−1. DNA was then extracted from these bacterial concentrations and fragmented, generating various amounts of the specific 227 bp dsDNA target. Following the same assay protocol, fragmented genomic DNA was added into reaction tubes containing capture probe-functionalized beads and padlock primers. Tubes were rinsed thoroughly after the target hybridization step to remove unbound DNA, as the vast majority of the fragmented genomic DNA is unable to hybridize with the designed capture probes or padlock primers. Captured targets were amplified as before, labeled with fluorescent probes, and imaged. Similar to the PCR-generated target results, the target generated directly from extracted genomic DNA was detected by the assay. The amount of counted target molecules has a linear trend on a double logarithmic plot down to a starting bacteria concentration of 1000 cfu ml−1 (Fig. 3). Our developed assay was able to detect the presence of genomic M. tb.DNA in concentrations as low as 10000 cfu ml−1, which could potentially enable the direct detection ofM. tb. bacteria in clinical samples.31
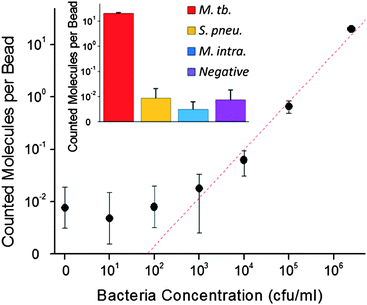 | ||
| Fig. 3 Number of detected molecules per bead is plotted as a function of initial bacteria concentration on a double-logarithmic scale. The relationship is linear (R2 = 0.991) to 103 cfu ml−1. Error bars are ± 1 s.d. (n = 6). Inset, specificity experiment; number of detected molecules per bead from M. tb. (red) Streptococcus pneumoniae (yellow), and Mycobacterium intercellulare (blue), and a negative control (no DNA, purple) are plotted. Error bars are ± 1 s.d. (n = 6). | ||
Experiments were also performed to test the specificity of our M. tb. detection assay, described in the ESI.†Genomic DNA samples from Streptococcus pneumoniae and Mycobacterium intercellulare were prepared and detected by following the same procedure as those used for M. tb. samples. The number of detected molecules per bead from the different bacteria is shown (Fig. 3, inset). The number of detected molecules per bead for M. tb. is significantly higher than the other two bacteria for similar concentrations of genomic DNA. S. pneumoniae and M. intracellulare do not contain the IS6110 region, and therefore should not be detected. Indeed, the signals from these bacteria samples are similar to a negative control in which no genomic DNA was presented to the assay. The results confirm the specificity of our M. tb. assay and also show that other non-tuberculosis mycobacterium are not detected, unlike traditional acid fast bacilli (AFB) smears.
Currently, the most sensitive detection of tuberculosis is through culturing of the bacteria. Unfortunately, this can take anywhere from 14 to 21 or more days,32 making it a less-than-ideal choice for rapid detection. Culturing is usually combined with a medical exam including chest X-rays and a medical history evaluation. A less sensitive but much faster alternative is a microscope analysis of smeared sputum samples to look for individual bacteria. Traditionally, the decision to place individuals with suspected TB in respiratory isolation is based upon the presence of positive or negative AFB smears of respiratory samples for M. tb. An average concentration of 1000 cfu ml−1 is generally required for an individual's respiratory sample to smear positive. Unfortunately, AFB smears do not distinguish between tuberculosis and infections with other non-tuberculosis mycobacteria. However, our designed assay presented here is able to distinguish between M. tb. and non-tuberculosis mycobacterium and, with further optimization, detect M. tb. near the required sensitivity of 1000 cfu ml−1. More importantly, the assay can give a positive diagnosis in a much shorter time than culturing.
In the developed detection assay presented here, amplified molecules were detected by the counting software even when no target molecules were present. We believe this could be a result of two processes. First, aggregations of fluorescently labeled oligonucleotides could generate a localized bright spot, leading to a detected amplified molecule. Second, it has been reported that certain polymerases can synthesize DNA from solutions containing no template or primer oligonucleotides.33 If this spontaneously synthesized DNA were to non-specifically hybridize with circular primers, they would be extended identically to amplified target molecules.23 As mentioned before, only one round of amplification is needed, which limits the amount of amplified “noise” in the assay. It is important to note that some amplified molecules are counted in the negative controls. If used in a clinical setting, a threshold (similar to a threshold cycle for PCR) would need to be set to ensure that non-specific amplification or detection would result in a false positive diagnosis.
Many experimental parameters affect the detection limit of the assay. Initially, a rapid cooling to room temperature was performed after the dsDNA target denaturing step. When the cooling was slowed down and the solution was maintained at 50 °C for a short time, a dramatic increase in counted target molecules was observed. Of equal importance is the incubation time of the denatured dsDNA target, the capture beads, and the padlock primers. When this was allowed to proceed for over 16 h, the number of counted target molecules was increased by an order of magnitude over a short, 2 min incubation time. An incubation time this long has been reported for increasing the sensitivity of DNA hybridization assays.11,34 Finally, these steps were combined for a final, optimized effect. After dsDNA target denaturing at 95 °C, the solution was cooled slowly to 50 °C and held at that temperature overnight, allowing for a maximum number of target molecules to be captured on the surface of the beads.
4. Conclusion
In conclusion, we have developed an assay for the detection of double-stranded DNA targets based on rolling circle amplification. The sensitivity of the developed method for dsDNA targets is a significant improvement to previously reported assays in literature, allowing detection down to 4.25 fM. Our assay shows specific genomic DNA detection of the pathogen Mycobacterium tuberculosis in bacterial concentrations of 10000 cfu ml−1, enabling the potential detection ofM. tb. bacteria in clinical samples. While the sensitivity of the assay is high, the speed of the assay is currently its biggest drawback. The assay's speed could be improved by utilizing a microfluidic chip, similar to that used by Sato and coworkers.35 By using a microfluidic channel to physically entrap beads, they showed a similar detection level of padlock probes as our method has detected dsDNA targets (500 zmol versus our 425 zmol), but with a greatly reduced target incubation time: only four hours versus our two days.
We are also looking to improve the sensitivity of the assay towards extracted genomic DNA targets, especially actual patient samples. By removing the current purification step needed before restriction enzyme digestion, membrane filters would not be needed, which eliminates any DNA losses due to permanent adsorption. Multiplex detection for a variety of pathogens could be achieved in the same assay by designing specific capture and padlock probes for different targets; amplified targets could subsequently be labeled and counted with separate fluorescent oligonucleotides. Finally, changing the assay to electrochemical detection36 would allow this assay to be performed on an electrode, eliminating the need for a fluorescence microscope setup.
Acknowledgements
This work was financially supported by the Bill and Melinda Gates Foundation.References
- C. G. Ban, S. M. Chung, D. S. Park and Y. B. Shim, Nucleic Acids Res., 2004, 32, e110 CrossRef.
- T. T. Goodrich, H. J. Lee and R. M. Corn, J. Am. Chem. Soc., 2004, 126, 4086–4087 CrossRef CAS.
- N. Li, J. S. Li and W. W. Zhong, Electrophoresis, 2008, 29, 424–432 CrossRef CAS.
- P. M. Lizardi, X. H. Huang, Z. R. Zhu, P. Bray-Ward, D. C. Thomas and D. C. Ward, Nat. Genet., 1998, 19, 225–232 CrossRef CAS.
- V. C. Martins, F. A. Cardoso, J. Germano, S. Cardoso, L. Sousa, M. Piedade, P. P. Freitas and L. P. Fonseca, Biosens. Bioelectron., 2009, 24, 2690–2695 CrossRef CAS.
- J. Wang, X. H. Cai, G. Rivas, H. Shiraishi and N. Dontha, Biosens. Bioelectron., 1997, 12, 587–599 CrossRef CAS.
- Z. P. Li, W. Li, Y. Q. Cheng and L. T. Hao, Analyst, 2008, 133, 1164–1168 RSC.
- X. J. Zhao, R. Tapec-Dytioco and W. H. Tan, J. Am. Chem. Soc., 2003, 125, 11474–11475 CrossRef CAS.
- J. E. Burton, J. Oshota, E. North, M. J. Hudson, N. Polyanskaya, J. Brehm, G. Lloyd and N. J. Silman, Mol. Cell. Probes, 2005, 19, 349–357 CrossRef CAS.
- T. Kostic, A. Weilharter, S. Rubino, G. Delogu, S. Uzzau, K. Rudi, A. Sessitsch and L. Bodrossy, Anal. Biochem., 2007, 360, 244–254 CrossRef CAS.
- D. Y. Lee, K. Shannon and L. A. Beaudette, J. Microbiol. Methods, 2006, 65, 453–467 CrossRef CAS.
- G. Panicker, D. R. Call, M. J. Krug and A. K. Bej, Appl. Environ. Microbiol., 2004, 70, 7436–7444 CrossRef CAS.
- N. Zammatteo, L. Jeanmart, S. Hamels, S. Courtois, P. Louette, L. Hevesi and J. Remacle, Anal. Biochem., 2000, 280, 143–150 CrossRef CAS.
- A. Almadidy, J. Watterson, P. A. E. Piunno, S. Raha, I. V. Foulds, P. A. Horgen, A. Castle and U. Krull, Anal. Chim. Acta, 2002, 461, 37–47 CrossRef CAS.
- H. Kuhn and M. D. Frank-Kamenetskii, Nucleic Acids Res., 2008, 36, e40 CrossRef.
- A. A. Gorodetsky, A. Ebrahim and J. K. Barton, J. Am. Chem. Soc., 2008, 130, 2924–2925 CrossRef CAS.
- K. Yoshitake, S. Waki and H. Ueda, Biosens. Bioelectron., 2008, 23, 1266–1271 CrossRef CAS.
- M. Minunni, S. Tombelli, J. Fonti, M. M. Spiriti, M. Mascini, P. Bogani and M. Buiatti, J. Am. Chem. Soc., 2005, 127, 7966–7967 CrossRef CAS.
- G. A. Blab, T. Schmidt and M. Nilsson, Anal. Chem., 2004, 76, 495–498 CrossRef CAS.
- J. Jarvius, J. Melin, J. Goransson, J. Stenberg, S. Fredriksson, C. Gonzalez-Rey, S. Bertilsson and M. Nilsson, Nat. Methods, 2006, 3, 725–727 CrossRef CAS.
- F. Dahl, J. Baner, M. Gullberg, M. Mendel-Hartvig, U. Landegren and M. Nilsson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4548–4553 CrossRef CAS.
- D. A. Di Giusto, W. A. Wlassoff, J. J. Gooding, B. A. Messerle and G. C. King, Nucleic Acids Res., 2005, 33, e64 CrossRef.
- T. Murakami, J. Sumaoka and M. Komiyama, Nucleic Acids Res., 2009, 37, e19 CrossRef.
- G. Nallur, C. H. Luo, L. H. Fang, S. Cooley, V. Dave, J. Lambert, K. Kukanskis, S. Kingsmore, R. Lasken and B. Schweitzer, Nucleic Acids Res., 2001, 29, 118e CrossRef , art. no.-e118.
- E. Schopf, N. O. Fischer, Y. Chen and J. B. H. Tok, Bioorg. Med. Chem. Lett., 2008, 18, 5871–5874 CrossRef CAS.
- N. R. Markham and M. Zuker, Nucleic Acids Res., 2005, 33, W577–W581 CrossRef CAS.
- G. T. Noordhoek, A. H. J. Kolk, G. Bjune, D. Catty, J. W. Dale, P. E. M. Fine, P. Godfreyfaussett, S. N. Cho, T. Shinnick, S. B. Svenson, S. Wilson and J. D. A. Vanembden, J. Clin. Microbiol., 1994, 32, 277–284 CAS.
- M. J. Torres, A. Criado, J. C. Palomares and J. Aznar, J. Clin. Microbiol., 2000, 38, 3194–3199 CAS.
- D. Thierry, M. D. Cave, K. D. Eisenach, J. T. Crawford, J. H. Bates, B. Gicquel and J. L. Guesdon, Nucleic Acids Res., 1990, 18, 188–188 CrossRef CAS.
- J. Li, C. S. H. Young, P. M. Lizardi and D. F. Stern, Cell Cycle, 2005, 4, 1599–1773 CrossRef CAS.
- F. S. Nolte, B. Metchock, J. E. McGowan, A. Edwards, O. Okwumabua, C. Thurmond, P. S. Mitchell, B. Plikaytis and T. Shinnick, J. Clin. Microbiol., 1993, 31, 1777–1782 CAS.
- F. A. Drobniewski, M. Caws, A. Gibson and D. Young, Lancet Infect. Dis., 2003, 3, 141–147 CrossRef CAS.
- N. Ogata and T. Miura, Nucleic Acids Res., 1998, 26, 4652–4656 CrossRef CAS.
- J. A. Ferguson, F. J. Steemers and D. R. Walt, Anal. Chem., 2000, 72, 5618–5624 CrossRef CAS.
- K. Sato, A. Tachihara, B. Renberg, K. Mawatari, K. Sato, Y. Tanaka, J. Jarvius, M. Nilsson and T. Kitamori, Lab Chip, 2010, 10, 1262–1266 RSC.
- L. Zhou, L. J. Ou, X. Chu, G. L. Shen and R. Q. Yu, Anal. Chem., 2007, 79, 7492–7500 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, M. tb. culturing, M. tb.genomic DNA extraction, M. tb.sample preparation, software algorithm, negative control experiments. See DOI: 10.1039/c0ay00529k |
| ‡ Current address: School of Pharmacy and Pharmaceutical Sciences, Departments of NanoEngineering and of Materials Science and Engineering, University of California at San Diego, La Jolla, California 92093, USA. |
| This journal is © The Royal Society of Chemistry 2011 |