DOI:
10.1039/C0AN00171F
(Paper)
Analyst, 2011,
136, 98-106
Systematic prediction error correction: A novel strategy for maintaining the predictive abilities of multivariate calibration models
Received
24th March 2010
, Accepted 21st September 2010
First published on 14th October 2010
Abstract
The development of reliable multivariate calibration models for spectroscopic instruments in on-line/in-line monitoring of chemical and bio-chemical processes is generally difficult, time-consuming and costly. Therefore, it is preferable if calibration models can be used for an extended period, without the need to replace them. However, in many process applications, changes in the instrumental response (e.g. owing to a change of spectrometer) or variations in the measurement conditions (e.g. a change in temperature) can cause a multivariate calibration model to become invalid. In this contribution, a new method, systematic prediction error correction (SPEC), has been developed to maintain the predictive abilities of multivariate calibration models when e.g. the spectrometer or measurement conditions are altered. The performance of the method has been tested on two NIR data sets (one with changes in instrumental responses, the other with variations in experimental conditions) and the outcomes compared with those of some popular methods, i.e. global PLS, univariate slope and bias correction (SBC) and piecewise direct standardization (PDS). The results show that SPEC achieves satisfactory analyte predictions with significantly lower RMSEP values than global PLS and SBC for both data sets, even when only a few standardization samples are used. Furthermore, SPEC is simple to implement and requires less information than PDS, which offers advantages for applications with limited data.
1. Introduction
Development of a multivariate calibration model for on-line/in-line process spectroscopy can sometimes be difficult and time-consuming because it involves several steps including selection and preparation of a large number of standard samples, measurement of spectra, and development and validation of a calibration model. Therefore, it is preferable if the calibration models can be used for an extended period. However, there are many situations in which a multivariate calibration model can become invalid.1,2 For example, changes in the physical (e.g. viscosity, particle size, surface tension) and/or chemical (e.g. starting materials in batch processes) characteristics of samples can lead to strongly biased predictions from multivariate calibration models. Changes can also be introduced by alterations to equipment that affect the instrumental responses measured, e.g. when a calibration model developed on one instrument has to be applied to spectra collected on another instrument, or when the instrumental response of a single instrument is subject to variations caused by either instrument ageing or repair. Another situation that may cause multivariate calibration models to be inapplicable is a variation in measurement conditions such as temperature, which can cause non-linear shifts and broadening of spectral absorption bands in multi-component samples owing to changes in intermolecular forces.3,4 Temperature changes may also introduce thermal expansion in instrument units affecting the alignment of optical components and cause peaks to shift along the wavelength axis.5
When spectral measurements are subject to the changes and variations mentioned above, methods for calibration model maintenance1,2,6–19 are needed to prevent degradation in the accuracy and reliability of multivariate calibration models and avoid time-consuming full recalibration procedures. Calibration model maintenance methods can be roughly classified into three categories, i.e. calibration model coefficient updating methods,6–8 prediction correction methods9–11 and spectral response standardization methods.12–19 In the literature, some wavelength selection and spectral pre-processing methods such as genetic algorithm (GA),20 simulated annealing (SA),21 finite impulse response (FIR),22 multiplicative signal correction (MSC),23–25 orthogonal signal correction (OSC),26,27 wavelets,28 external parameter orthogonalization,29 orthogonal projection30 and dynamic orthogonal projection31 have also been used to address the same issue. Since they should be applied before the establishment of multivariate calibration models, they are not calibration model maintenance methods in essence.
Calibration model coefficient updating involves recalculation of the model coefficients (e.g. regression vector) with the addition of a few new samples to the old calibration set. Model coefficient updating can generally be applied to systems where spectral differences induced by the changes or variations in instrument or measurement conditions are not too complicated. But for complex situations, a substantial number of samples would be required to obtain satisfactory results.
One of the most widely used methods for correcting predicted values is the simple univariate slope and bias correction (SBC) method.9 Since SBC is a univariate approach, it works well when the spectral differences induced by the changes in instrument or measurement conditions are simple and systematic for all samples. If such spectral differences are more complex and differ from sample to sample, the applicability of SBC will be questionable.
The idea of spectral response standardization is to find a transformation matrix, which transforms the spectra of future test samples into the corresponding spectra as if they were measured under the same conditions or on the same instrument as the calibration samples used to build the original calibration model. Therefore, the original calibration model can be used for prediction without having to update the model coefficients. The transformation matrix can be obtained by regressing the spectra of a subset of samples (often called standardization samples) measured on the primary instrument (or under initial calibration conditions) against the spectra of the same subset measured on the secondary instrument (or under the modified test conditions). Among the existing spectral response standardization methods, piecewise direct standardization (PDS) is probably the most widely used and discussed method.14,32–38 PDS can handle complex situations due to its multivariate character. In PDS, the transformation matrix is estimated by the moving window procedure, which enables better modeling of possible nonlinearities. However, the window size has a significant effect on the performance of PDS and needs to be carefully determined. Furthermore, with PDS, the spectra of the standardization samples must be measured on both instruments or under both sets of measurement conditions, which makes the procedure inapplicable to on-line/in-line monitoring of complex chemical and bio-chemical processes where such a requirement is difficult to satisfy in practice.
Due to various theoretical and practical limitations of the existing calibration model maintenance methods, there is still a need for methods that are easier to implement (i.e. require fewer or even no meta-parameters) and at the same time provide better performance. In this paper, a new strategy for calibration model maintenance is described and its performance is compared to that of PDS, SBC and global PLS using two NIR data sets.
2. Systematic prediction error correction (SPEC)
Suppose a multivariate calibration model, c = f(x) has been established on the spectra (Xcal) of a set of calibration samples measured at the selected calibration conditions or on the primary instrument (x and c represent the spectrum of a sample and the concentration of the target constituent in the sample, respectively). The task is now to enable the calibration model to give correct quantitative predictions for the target constituent in test samples, based on their spectra measured at the test conditions or on a secondary instrument.
Assume the rows of the m × p spectral matrices X1 and X2 are the corresponding spectra at p wavelengths of the same subset of m standardization samples measured under the calibration and test conditions (or on the primary and secondary instruments), respectively. According to the Beer–Lambert law, X1 and X2 can be decomposed as follows:
| | | X1 = CST1 + E1; X2 = CST2 + E2 | (1) |
where, superscript ‘T’ denotes the transpose.
C is the
m ×
r concentration
matrix with its
i-th row representing the concentrations of all the chemical components in the
i-th standardization sample (
r is the actual number of spectroscopically active chemical components in the samples).
S1 and
S2 are pure spectral matrices with size of
p ×
r, whose columns are the pure spectra of chemical components in the standardization samples at the calibration and test conditions (or on the primary and secondary instruments), respectively.
E1 and
E2 denote the corresponding residual matrices.
If S1 and S2 are known a priori, for a test sample, its spectrum (xtest) measured at the test conditions (or on the secondary instrument) can be easily transformed into the spectrum (xtrans) as if it were measured at the calibration conditions (or on the primary instrument) through the simple calculations in eqn (2). The multivariate calibration model built at the calibration conditions (or on the primary instrument) can then be used to predict the concentration of the target constituent in the test sample from the transformed spectrum,
| | 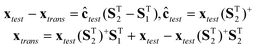 | (2) |
where ‘+’ symbolizes the Moore–Penrose generalized inverse.
In most cases, it is difficult, if not impossible, to obtain S1 and S2. Fortunately, for an r × r full rank matrix R, the following equations hold.
| | | (RST2)+RST1 = (ST2)+R+RST1 = (ST2)+ST1 | (3) |
| | | (RST2)+RST2 = (ST2)+R+RST2 = (ST2)+ST2 | (4) |
Therefore, (ST2)+ST1 and (ST2)+ST2 in eqn (2) can be replaced by (RST2)+RST1 and (RST2)+RST2, respectively, which are readily obtained by singular value decomposition of Xcomb (Xcomb = [X1,X2]),
| | 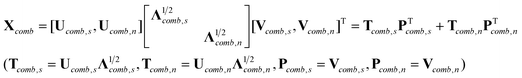 | (5) |
where subscripts ‘
s’ and ‘
n’ signify that the corresponding factors represent spectral information and noise, respectively.
Pcomb,s consists of
r columns. We then partition
PTcomb,s into two submatrices
PT1 and
PT2 (
PTcomb,s = [
PT1,
PT2]), which have the same sizes as
ST1 and
ST2, respectively. Since
Xcomb = [
X1,
X2] =
C·[
ST1,
ST2] +
E, it is easy to prove that there exists an
r ×
r full rank
matrix R satisfying the following equations.
| | RST1 = PT1,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) RST2 = PT2 RST2 = PT2 | (6) |
Combining eqn (2), (3), (4) and (6), one can get
| | | xtrans = xtest(PT2)+PT1 + xtest − xtest(PT2)+PT2 | (7) |
The above transformation method is a special case of the loading space standardization method developed by some of the present authors.39 The concentration of the target constituent in the test sample (ĉtest) can be predicted from its standardized spectrum (xtrans) by the multivariate linear calibration model (c = f(x) = a + xb) established on the spectra (Xcal) of calibration samples measured at the calibration conditions (or on the primary instruments).
| | 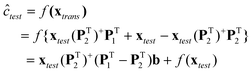 | (8) |
Obviously, the applicability of eqn (8) lies in the availability of the spectra (X1 and X2) of a subset of standardization samples recorded at both the calibration and test conditions (or on both the primary and secondary instruments). However, such a requirement is difficult to satisfy in the area of on-line/in-line monitoring of chemical, bio-chemical and pharmaceutical processes. In such applications, it is relatively easier to measure the spectra of a subset of standardization samples (X2) at the test conditions (or on the secondary instrument), and the concentrations of the target constituent (c2) in the standardization samples through off-line assay. Under such circumstances, P1 is not available; hence eqn (8) is not directly applicable. Nevertheless, introducing [I − (PT1)+PT1] (I is an identity matrix of appropriate size) into eqn (8), gives the following equation.
| | 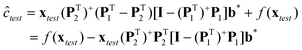 | (9) |
Since the rows of Xcal and the unavailable X1 of a subset of standardization samples should theoretically span the same spectral space, (PT1)+PT1 in the above equation can then be replaced by (PTcal,s)+PTcal,s, which can be obtained by singular value decomposition of Xcal.
| | 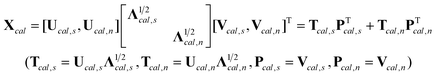 | (10) |
With
X2 and
c2,
b*can be readily estimated out by PLS using mean-centring from
eqn (11) (the number of principal components used in PLS can be set to
r − 1).
| | | f(X2) − c2 = X2(PT2)+PT2[I − (PTcal,s)+PTcal,s]b* | (11) |
For a test sample, the concentration of the target constituent can therefore be directly calculated from its spectrum (xtest) measured at the test conditions or on the secondary instrument without spectral transformation.
| | | ĉtest = f(xtest) − xtest(PT2)+PT2[I − (PTcal,s)+Pcal,s]b* | (12) |
In eqn (12), the term, xtest(PT2)+PT2[I − (PTcal,s)+Pcal,s]b*, can be regarded as the systematic prediction error of the multivariate linear calibration model caused by spectral differences resulting from variations in measurement conditions or changes in instrument. This is why the above method for calibration model maintenance is called Systematic Prediction Error Correction (SPEC). It is worthwhile pointing out that although the above method is deduced under the assumption of Beer–Lambert law, it can be applied as an efficient linear approximation to situations where the Beer–Lambert law does not strictly hold.
3. Applications
3.1
NIR data of
pharmaceutical
tablets
This data is available at http://www.idrc-chambersburg.org/shootout2002.html. It consists of 1308 spectra of 655 pharmaceutical tablets measured on two Multitab spectrometers (FossNIRsystems, Silverspring, MD) in the transmittance mode from 600 to 1898 nm in 2 nm increments. Each individual tablet was subsequently analyzed for assay value of the active ingredient, tablet weight, and tablet hardness. For each of the 1308 absorbance spectra, the 520 absorbance values in the range between 600 and 1638 nm were used in the subsequent data analysis. Absorbance spectra from each instrument were split into a calibration set (155 spectra), a test set (460 spectra) and a validation set (40 spectra). The calibration set includes tablets with a wide range of assay values (151.6–239.1 mg). The challenge for this data set is to develop a multivariate linear calibration model for the assay value of the active ingredient on one instrument (the primary instrument), and then to provide the best means of transferring the calibration model to the other instrument (the secondary instrument). Several possible outliers (calibration set: no.19, 122, 126, and 127; the test set: no.11, 145, 267, 295, 294, 342, 313, 341 and 343) were excluded from the subsequent analysis. Therefore, the actual number of samples in the calibration set and the test set were 151 and 451, respectively.
3.2
NIR data of
fermentation
process
The process under study was an industrial pilot-plant scale streptomyces fermentation in a 12 litre vessel involving a seed stage and a production stage. Biomass was grown in the seed stage and then transferred into the final stage for the production of the product. The production stage was a fed batch process lasting approximately 140 h. Two sets of fermentation experiments were carried out. The first set comprised seven batches (referred to as ‘calibration batches’). And the second set was made up of six batches (referred to as ‘test batches’). The seven calibration batches were run under similar conditions (see ref. 37 for details), but natural variation provided a degree of variability in the resulting data. For the test batches, the experiments were carried out at different environmental conditions (pH and temperature), feed rates (sugar feed) and feed amounts (oil feed). A summary of the experimental conditions of the test batches is given in Table 1. NIR measurements were collected on-line from the production stage of all the batches. NIR spectra between 950 and 1700 nm were recorded with a resolution of 6 nm every fifteen minutes using a Zeiss Corona 45 NIR (Carl Zeiss, Germany) operated in reflectance mode. An integration time of 17 ms was employed and 10 scans were accumulated for each spectrum; hence, the time for acquisition of one spectrum was 170 ms. Log(1/R) spectra between 1064 and 1430 nm were selected for the subsequent data analysis. For the detailed experimental setup, refer to ref. 40. The key parameter considered in the analysis was product concentration in the broth. The concentration of the product was determined by off-line analysis using HPLC with around ten samples taken throughout the duration of the batch. The aim of the analysis was to investigate the predictive performance of the spectral calibration model built on the calibration batches for the test batches, when different calibration model maintenance methods are applied.
Table 1 The experimental conditions of the test fermentation batches
| Test Batch |
T/°C |
pH |
Sugar (g hr−1) |
Oil (%) |
|
there was a leak in the oil feed vessel and hence, the reaction was carried out with a low oil content.
|
| 1 |
25 |
6.8 |
9.8 |
lowa |
| 2 |
23 |
6.5 |
10.8 |
4.6 |
| 3 |
27 |
7.1 |
10.8 |
4.6 |
| 4 |
27 |
6.5 |
10.8 |
3.7 |
| 5 |
27 |
6.5 |
8.8 |
4.6 |
| 6 |
23 |
6.5 |
8.8 |
3.7 |
3.3
Data analysis
Data pre-treatment.
No further data pre-treatment other than the outlier rejection and wavelength region selection as stated above was applied to the pharmaceutical tablet data. For the NIR data of the fermentation processes, the spectra were first preprocessed to the first derivative using the Savitsky–Golay method41 with a 9-point window and a second-order polynomial, and then each first-derivative spectrum was normalized through dividing by its l2-norm. The third step in the pre-processing stage was a consequence of the product concentrations being measured by off-line assay and thus the number of values obtained during a fermentation was restricted to about ten. For each batch, a second-order polynomial was fitted between the sampling time and the product concentration determined by off-line assay. The values of the product concentration at the same sampling times as the spectral measurements were obtained by interpolation. The dynamics of the process were sufficiently slow to justify such an interpolation strategy.
The optimal calibration models.
The PLS regression method (PLS_Toolbox3.0, Eigenvector Research Inc.) was selected to build the calibration models for both the pharmaceutical tablet data and fermentation data, using mean-centring. The optimal PLS calibration model for the pharmaceutical tablet data built on the spectra of the calibration set measured on the primary instrument was chosen to be the one with minimal RMSEP value for the test set. Due to the relatively lower number of batches in the calibration set of fermentation data, the optimal PLS calibration model for the fermentation data established on the spectra of the calibration batches was determined by leave-one-batch-out cross validation.
The standardization samples.
For the pharmaceutical tablet data, the standardization samples used in calibration model maintenance were chosen from the calibration samples by first randomly selecting a sample and then sequentially adding a new sample which carries the most new spectral information not contained in the samples already selected, until the number of samples reaches the predefined value. Although the choice of the first standardization sample led to slightly different results, it did not alter the order of the performance of the calibration model maintenance methods discussed herein (i.e. SPEC, PDS, SBC and global PLS). Therefore, sample no.120 was arbitrarily chosen from the corresponding calibration sets as the first standardization sample for the pharmaceutical tablet data. For the fermentation data, the standardization spectral matrix (X2) for each test batch consisted of 70 successive NIR spectra starting with the spectrum measured at the same time as the first offline assay, and the corresponding concentration vector (c2) was composed of the interpolation concentrations and two to four offline assay values.
Some aspects of the implementation of calibration model maintenance methods.
The optimal global PLS calibration model built on the spectral data assembled by the calibration samples and standardization samples was determined by leave-one-out cross validation. The transformation matrix of PDS for the pharmaceutical tablet data was estimated from the spectra of the standardization samples by the PLS method using mean-centring. It should be pointed out that PDS could not be applied to the fermentation data due to the unavailability of the standardization spectral matrix (X1), the counterpart of the standardization spectral matrix (X2) for each test batch. The singular value decomposition in SPEC was carried out on the raw spectra of the standardization samples. The root-mean-square error of prediction (RMSEP) was used as the performance criterion to assess the performance of SPEC, PDS, SBC and global PLS for calibration model maintenance.| | 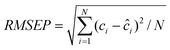 | (13) |
Here, ci is the known concentration of the target constituent in the ith sample, ĉi denotes the corresponding value estimated from the corresponding calibration models, and N denotes the number of samples. All the programmes for calibration model maintenance were written in house using Matlab6.5 (Mathworks Inc.).
4. Results and discussions
4.1
The
pharmaceutical
tablet data
4.1.1 Effects of changes in instrumental responses
Although the spectra of the same sample measured using two different instruments of the same type may have the same basic shape or profile, subtle differences in instrumental response functions can result in perceptible spectral variations. This is illustrated in Fig. 1, where significant spectral variations can be observed in the region between 600 and 720 nm; there are also subtle spectral differences in other regions. These spectral variations can cause large systematic prediction errors when the calibration model built on the primary instrument is applied to the spectra measured on the secondary instrument. As shown in Fig. 2, the differences in instrumental responses resulted in significant offsets in the prediction of the active mass with the secondary instrument, when a PLS calibration model built using the primary instrument was applied. Besides affecting accuracy, the change in instrument also degraded the predictive precision of the calibration model, which signifies that the spectral variations introduced by differences between the instruments are not systematic for all samples, but rather differ from sample to sample.
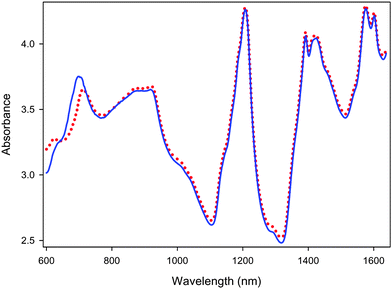 |
| | Fig. 1
Spectra of the same pharmaceutical tablet obtained with the primary (red dotted line) and secondary (blue solid line) instruments. | |
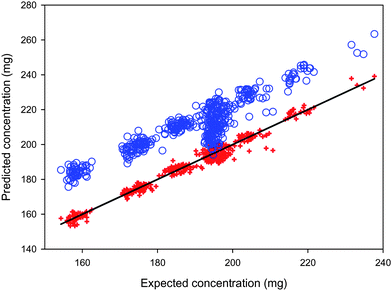 |
| | Fig. 2 Mass of the active ingredient in the test pharmaceutical tablet samples predicted from their spectra recorded with the primary (red cross) and the secondary instruments (blue circle) using a PLS calibration model with eleven principal components built from the calibration spectra obtained using the primary instrument. Diagonal line: theoretically correct predictions. | |
4.1.2 Model parameters of the calibration model maintenance methods
Four methods (SPEC, PDS, SBC and global PLS) were used to eliminate the detrimental effects of instrumental changes with a view to maintaining the predictive abilities of the calibration model. Before the application of these methods, the influence of some model parameters on their performance was investigated: the number of standardization samples and the number of principal components, and the window size (exclusively for PDS).
The number of standardization samples.
In order to obtain reliable results, the number of standardization samples should be at least equal to the number of actual spectral variation sources in the calibration spectra. Of course, the larger the number of standardization samples, the higher the probability that good results will be obtained. However, more standardization samples require increased analysis time and effort, with associated higher costs. Therefore, a calibration maintenance method that achieves satisfactory results with fewer standardization samples is preferred in practice. The effects of the number of standardization samples on the performances of the four calibration maintenance methods (SPEC, PDS and SBC, global PLS) are shown in Fig. 3. The main observation is that SPEC with only a few standardization samples can obtain predictions with quite low RMSEP values. Further increasing the number of standardization samples does not significantly reduce the RMSEP values. Compared to PDS, SBC and global PLS with the same number of standardization samples, SPEC can generally provide predictions with lower RMSEP values. In order to obtain predictions with comparable RMSEP values, PDS, SBC and global PLS need more standardization samples than SPEC does. Overall, SPEC is more efficient than the other three methods in maintaining the accuracy and reliability of calibration models.
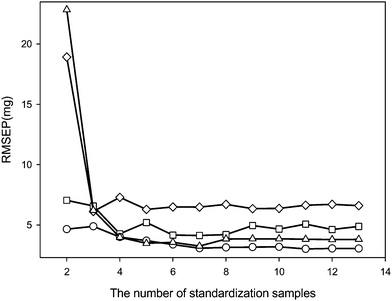 |
| | Fig. 3 The effects of the number of standardization samples on the performance of four calibration maintenance methods (circle: SPEC, triangle: PDS, square: global PLS, diamond: SBC). RMSEP values were calculated for the active ingredient mass in the pharmaceutical tablet samples (excluding the standardization samples) predicted from their spectra recorded with the secondary instrument. | |
The number of principal components (Npc).
In PDS, the number of principal components (PCs) is involved in the calculation of the transformation matrices; while in SPEC, it is related to the number of factors representing spectral information retained in P2 and Pcal,s after singular value decomposition of X2 and Xcal, respectively. Theoretically, the number of PCs used in SPEC should not be less than the number of actual spectral variation sources in the system under study. For PDS, the minimal number of PCs used in the calculation of the transformation matrices using PLS with mean-centring can be one less than the number of spectral variation sources. From Fig. 4a, it is seen that the RMSEP value of SPEC reaches its minimum (3.0 mg) when the first four principal components were included in P2 and Pcal,s. Increasing the number of PCs from four to seven has little effect on the performance of SPEC in terms of the RMSEP value. Furthermore, the range of RMSEP values for 2–12 PCs is only 3–4 mg. This is a useful attribute of SPEC as there is no need to estimate accurately the number of actual spectral variation sources in the spectra of the standardization samples. In practice, the number of PCs used in SPEC can be set to a value equal to or slightly larger than the number of significant singular values of Xcal. In contrast, the performance of PDS is greatly affected by the number of PCs used in the calculation of the transformation matrix. An inaccurate estimation of the number of actual spectral variation sources in the spectra of the standardization samples can lead to significant deterioration in the performance of PDS (Fig. 4b). Comparable values of RMSEP to those achieved by SPEC are obtained for 1–3 PCs, but thereafter the RMSEP increases significantly to over 50 mg when 12 PCs are used.
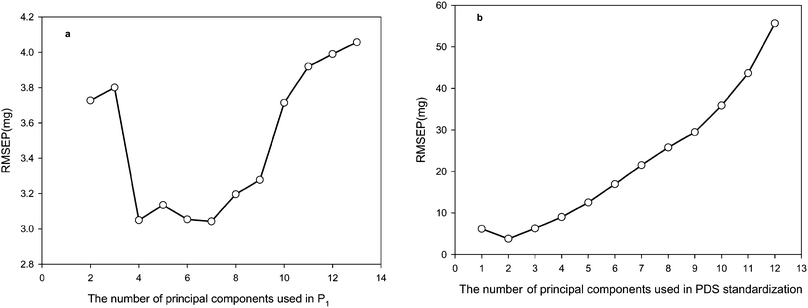 |
| | Fig. 4 a) The effects of the number of principal components retained in Pcal,s and P2 on the performance of SPEC with 13 standardization samples; b) the effects of the number of principal components used in the calculation of the transformation matrix on the performance of PDS with 13 standardization samples. RMSEP values were calculated for the active ingredient mass in the pharmaceutical tablet samples (excluding the standardization samples) from their spectra recorded with the secondary instrument. | |
Window size (Nws).
This parameter is only relevant to the PDS method. When the data for the pharmaceutical tablets were considered, it was found that the RMSEP value calculated for the active ingredient mass in the pharmaceutical tablet samples, from their spectra recorded with the secondary instrument, varied between 3.6 mg and 5.2 mg in an unsystematic manner when the window size was changed from 9 to 101 (not shown). The minimal RMSEP value was attained at a window size of 13. Therefore, when no extra spectra measured on the secondary instrument (or at the test conditions) except those of only a few standardization samples are available, it is difficult to determine the optimal window size for PDS.
4.1.3 The performance of the four model maintenance methods
Table 2 lists the RMSEP values for the active ingredient concentrations in the pharmaceutical tablet samples predicted by different models from their spectra measured with the secondary instrument. Without model maintenance, the PLS calibration model (PLS1) built on the spectra of the calibration samples measured with the primary instrument gave large errors in the predictions for the active ingredient mass in the tablet samples, based on the spectra measured with the secondary instrument. The RMSEP value for the entire pharmaceutical tablet data set is 22.7 mg, which is equivalent to an average relative predictive error of 11.7% (not shown in Table 2). The PLS model (PLSsub) built on the spectra of six standardization samples measured on the secondary instrument provided concentration predictions with substantially lower RMSEP values. However, compared with the fully recalibrated PLS2 model, the predictive errors of PLSsub are still moderately high.
Table 2
RMSEP values (mg) of the active ingredient concentrations in the tablet samples predicted by different methods from the spectra recorded with the secondary instrument (six standardization samples were used in global PLS, SBC, PDS and SPEC)abcd
| Models |
Calibration set |
Test set |
Validation set |
Entire set |
|
PLS1 denotes the PLS calibration model built using the spectra of the calibration samples recorded with the primary instrument.
PLSsub denotes the PLS calibration model built using the spectra of six standardization samples recorded with the secondary instrument.
PLSglobal signifies the global PLS model built using the spectra of the calibration samples recorded with the primary instrument and the six standardization samples recorded with the secondary instrument.
PLS2 represents the PLS calibration model built using the spectra of the calibration samples recorded with the secondary instrument.
|
|
PLS1 |
23.6 |
22.5 |
22.1 |
22.7 |
| PLSsub |
8.4 |
6.6 |
6.4 |
7.0 |
|
SBC-PLS1 |
5.7 |
6.6 |
7.6 |
6.5 |
| PLSglobal |
3.6 |
4.1 |
6.1 |
4.2 |
|
PDS-PLS1 |
3.4 |
3.2 |
6.7 |
3.6 |
|
SPEC-PLS1 |
3.6 |
3.1 |
5.3 |
3.4 |
|
PLS2 |
2.4 |
2.7 |
5.2 |
2.8 |
The application of calibration model maintenance methods with six standardization samples further brought down the predictive errors. Among the four calibration model maintenance methods, SBC offered only a slight improvement over PLSsub in terms of the RMSEP for the entire data set. It suggests that the spectral differences induced by the changes in instrument cannot be effectively modeled by the univariate approach of SBC. As expected, more sophisticate multivariate approaches, viz. global PLS, PDS (Npc: 2, Nws: 13) and SPEC (Npc: 4), achieved better results. In particular, the RMSEP values of SPEC and PDS compare favorably to those of the fully recalibrated PLS2 model. More interestingly, although SPEC utilized the same information as the global PLS procedure, lower RMSEP values were obtained, which demonstrated the effectiveness of the calibration maintenance strategy employed by SPEC.
4.2
Fermentation
data
Fermentation processes are rather complex. The variations in the environmental conditions (pH and temperature), feed rates (sugar feed) and feed amounts (oil feed) of the test batches could significantly deteriorate the predictive ability of the calibration model built on the calibration batches. Therefore, the successful application of spectroscopic process analytical technologies to the in situ monitoring of fermentation processes relies on the availability of effective calibration model maintenance methods. For the fermentation data, the performance of the three calibration model maintenance methods, SPEC, SBC and global PLS, were investigated and compared. From Table 3, it can be observed that the PLS calibration model established on the calibration batches could not provide satisfactory results for most of the test batches due to the differences in the experimental conditions. Though global PLS improved the quality of the predictions, its results were still not good enough for fermentation process control. SBC provided good results for tests batches 2, 4, 5 and 6, but for test batches 1 and 3 the results were inferior to those of the PLSsub models built on a few standardization samples. In comparison with global PLS, PLSsub and SBC, the performance of SPEC (Npc: 6) was generally better with SBC giving a somewhat significant lower RMSEP value for only test batch 5. The typical results displayed in Fig. 5 indicate the deficiencies of the global PLS and SBC models, and demonstrate the efficiency of SPEC in maintaining the predictive ability of the PLS1 model. It can be seen that changes in the fermentation conditions over time had almost no influence on the accuracy and reliability of the PLS1 model when SPEC was applied. The success in maintaining the predictive ability of the calibration models for a complex biochemical process further verified that SPEC is highly effective as a calibration model maintenance method for NIR spectrometry.
Table 3 The RMSEP values of the product concentrations in the test batches predicted by different methods from the corresponding near infrared spectra measured during the fermentation process
| Method |
Test 1 |
Test 2 |
Test 3 |
Test 4 |
Test 5 |
Test 6 |
|
PLS1 |
0.8062 |
0.2813 |
0.5844 |
0.0948 |
1.5994 |
1.7250 |
| PLSsub |
0.1222 |
0.2343 |
0.1532 |
0.2251 |
0.8646 |
0.0821 |
| PLSglobal |
0.1742 |
0.0969 |
0.4378 |
0.1465 |
0.2979 |
0.2535 |
|
SBC-PLS1 |
0.3139 |
0.1262 |
0.1730 |
0.1010 |
0.1815 |
0.0374 |
|
SPEC-PLS1 |
0.0435 |
0.0639 |
0.1493 |
0.1055 |
0.2303 |
0.0367 |
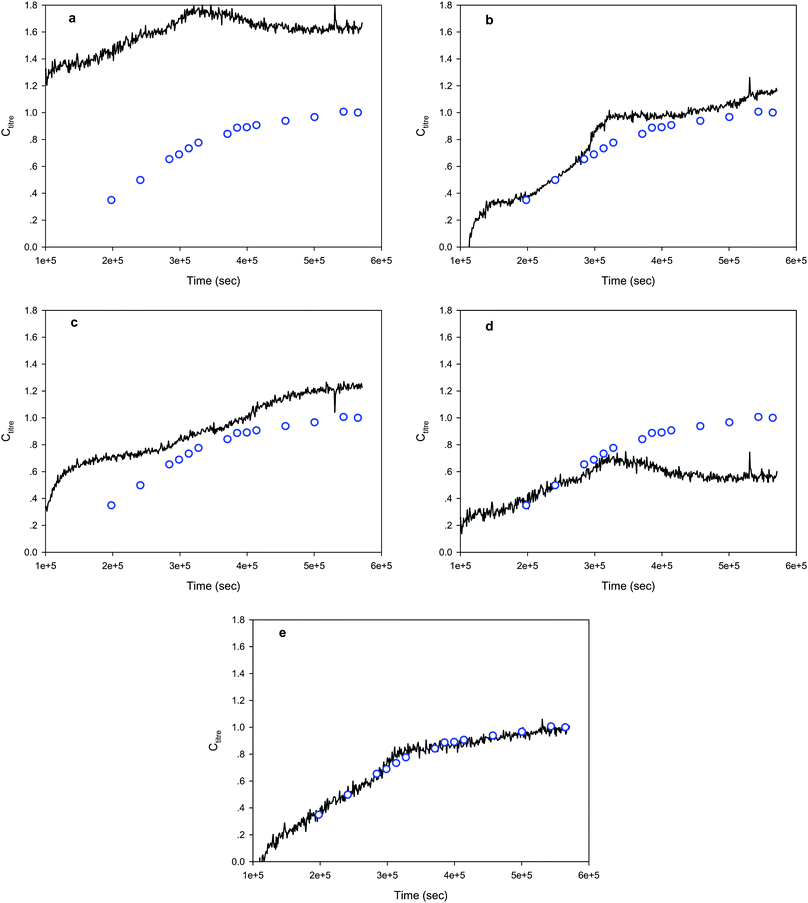 |
| | Fig. 5 The product concentration profiles (black solid lines) for one of the test fermentation batches predicted by different methods (a: PLS, b: PLSsub, c: PLSglobal, d: SBC, e: SPEC). The blue circles represent the product concentrations determined by off-line assay. | |
5. Conclusions
The novel calibration model maintenance method described, SPEC, significantly improved the predictive results of NIR calibration models for pharmaceutical tablet data and fermentation data affected by changes in instrument and experimental conditions, respectively. SPEC only requires the analyte concentrations in a few standardization samples and the corresponding spectra measured at the experimental conditions or on the instrument to which the calibration model is applied. Moreover, the implementation of SPEC is simple. Only one model parameter (i.e. the number of chemical variation sources in the spectral data) needs to be identified prior to the application of SPEC, which can be simply set to the number of the significant singular values of the spectral data. Consequently, SPEC has wider applicability than other standardization methods (e.g. PDS) that need the spectra of the standardization samples to be measured under both the calibration and test conditions or on both the primary and secondary instruments, and/or involve the tricky choice of the values of some meta-parameters without chemical meanings.
Acknowledgements
The authors thank National Nature Science Foundation of China (grant no. 21075034), CPACT and the University of Strathclyde Research Development Fund for support. DTI and EPSRC are thanked for the award of a LINK grant (KNOW–HOW)-(EPSRC grant no. GR/R19366/01). Clairet Scientific is also thanked for the loan of the Zeiss Corona spectrometer. AN acknowledges the award of a University Research Fellowship by the Royal Society, UK.
References
- E. Bouveresse and D. L. Massart, Vib. Spectrosc., 1996, 11, 3–15 CrossRef CAS.
- R. N. Feudale, N. A. Woody, H. Tan, A. J. Myles, S. D. Brown and J. Ferré, Chemom. Intell. Lab. Syst., 2002, 64, 181–192 CrossRef CAS.
- T. Iwata, J. Koshoubu, C. Jin and Y. Okubo, Appl. Spectrosc., 1997, 51, 1269–1275 CrossRef CAS.
- Y. Ozaki, Y. Liu and I. Noda, Appl. Spectrosc., 1997, 51, 526–535 CrossRef CAS.
- D. Ozdemir, M. Mosley and R. Williams, Appl. Spectrosc., 1998, 52, 1203–1209 CrossRef CAS.
- H. Swierenga, A. P. de Weijer and L. M. C. Buydens, J. Chemom., 1999, 13, 237–249 CrossRef CAS.
- C. V. Greensill, P. J. Wolfs, C. H. Spiegelman and K. B. Walsh, Appl. Spectrosc., 2001, 55, 647–653 CrossRef CAS.
- C. L. Stork and B. R. Kowalski, Chemom. Intell. Lab. Syst., 1999, 48, 151–166 CrossRef CAS.
- E. Bouveresse, C. Hartmann, D. L. Massart, I. R. Last and K. A. Prebble, Anal. Chem., 1996, 68, 982–990 CrossRef CAS.
- J. A. Jones, I. R. Last, B. F. MacDonald and K. A. Prebble, J. Pharm. Biomed. Anal., 1993, 11, 1227–1231 CrossRef CAS.
- E. Bouveresse, D. L. Massart and P. Dardenne, Anal. Chim. Acta, 1994, 297, 405–416 CrossRef CAS.
-
J. S. Shenk, M. O. Westerhaus, U.S. Patent 4866644, Sept 12, 1989.
- E. Bouveresse, D. L. Massart and P. Dardenne, Anal. Chem., 1995, 67, 1381–1389 CrossRef CAS.
- Y. D. Wang, D. J. Veltkamp and B. R. Kowalski, Anal. Chem., 1991, 63, 2750–2756 CrossRef CAS.
- F. W. Koehler, G. W. Small, R. J. Combs, B. B. Knapp and R. T. Kroutil, Anal. Chem., 2000, 72, 1690–1698 CrossRef CAS.
- F. Despagne, B. Walczak and D. L. Massart, Appl. Spectrosc., 1998, 52, 732–745 CrossRef CAS.
- R. Goodacre, E. M. Timmins, A. Jones, D. B. Kell, J. Maddock, M. L. Heginbothom and J. T. Magee, Anal. Chim. Acta, 1997, 348, 511–532 CrossRef CAS.
- D. T. Andrews and P. D. Wentzell, Anal. Chim. Acta, 1997, 350, 341–352 CrossRef CAS.
- Y. L. Xie and P. K. Hopke, Anal. Chim. Acta, 1999, 384, 193–205 CrossRef CAS.
- D. Ozdemir and R. Williams, Appl. Spectrosc., 1999, 53, 210–217 CrossRef CAS.
- H. Swierenga, P. J. de Groot, A. P. de Weijer, M. W. J. Derksen and L. M. C. Buydens, Chemom. Intell. Lab. Syst., 1998, 41, 237–248 CrossRef CAS.
- T. B. Blank, S. T. Sum, S. D. Brown and S. L. Monfre, Anal. Chem., 1996, 68, 2987–2995 CrossRef CAS.
- P. Geladi, D. MacDougall and H. Martens, Appl. Spectrosc., 1985, 39, 491–500.
- T. Isaksson and T. Naes, Appl. Spectrosc., 1988, 42, 1273–1284 CAS.
- C. F. Pereira, M. F. Pimentel, R. K. H. Galvāo, F. A. Honorato, L. Stragevitch and M. N. Martins, Anal. Chim. Acta, 2008, 611, 41–47 CrossRef CAS.
- J. Sjoblom, O. Svensson, M. Josefson, H. Kullberg and S. Wold, Chemom. Intell. Lab. Syst., 1998, 44, 229–244 CrossRef CAS.
- C. V. Greensill, P. J. Wolfs, C. H. Spiegelman and K. B. Walsh, Appl. Spectrosc., 2001, 55, 647–653 CrossRef CAS.
- B. Walczak, E. Bouveresse and D. L. Massart, Chemom. Intell. Lab. Syst., 1997, 36, 41–51 CrossRef CAS.
- J. Roger, F. Chauchard and V. Bellon-Maurel, Chemom. Intell. Lab. Syst., 2003, 66, 191–204 CAS.
- A. Andrew and T. Fearn, Chemom. Intell. Lab. Syst., 2004, 72, 51–56 CrossRef CAS.
- M. Zeaiter, J. M. Roger and V. Bellon-Maurel, Chemom. Intell. Lab. Syst., 2006, 80, 227–235 CrossRef CAS.
- H. Swierenga, W. G. Haanstra, A. P. de Weijer and L. M. C. Buydens, Appl. Spectrosc., 1998, 52, 7–16 CrossRef CAS.
- Y. Wang, M. J. Lysaght and B. R. Kowalski, Anal. Chem., 1992, 64, 562–564 CrossRef CAS.
- Y. Wang and B. R. Kowalski, Anal. Chem., 1993, 65, 1301–1303 CrossRef CAS.
- Z. Y. Wang, T. Dean and B. R. Kowalski, Anal. Chem., 1995, 67, 2379–2385 CrossRef CAS.
- H. W. Tan and S. D. Brown, J. Chemom., 2001, 15, 647–663 CrossRef CAS.
- E. Bouveresse and D. L. Massart, Chemom. Intell. Lab. Syst., 1996, 32, 201–213 CrossRef CAS.
- P. J. Gemperline, J. H. Cho, P. K. Aldridge and S. S. Sekulic, Anal. Chem., 1996, 68, 2913–2915 CrossRef CAS.
- Z. P. Chen, J. Morris and E. Martin, Anal. Chem., 2005, 77, 1376–1384 CrossRef CAS.
- A. Nordon, D. Littlejohn, A. S. Dann, P. A. Jeffkins, M. D. Richardson and S. L. Stimpson, Analyst, 2008, 133, 660–666 RSC.
- P. A. Gorry, Anal. Chem., 1990, 62, 570–573 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.