Well-defined star shaped polymer-fullerene hybrids via click chemistry†
Andrew J.
Inglis
a,
Philippe
Pierrat
b,
Thierry
Muller
b,
Stefan
Bräse
*b and
Christopher
Barner-Kowollik
*a
aPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstraße 18, 76128 Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu; Fax: +49 721 608 5740
bInstitut für Organische Chemie and Center for Functional Nanostructures (CFN), Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: stefan.braese@kit.edu.; Fax: +49 (0)721 608 8581; Tel: +49 (0)721-608-2902
First published on 30th October 2009
Abstract
The use of a hexakisazido macrocyclic methanofullerene proves to be highly efficient in the preparation of 6-arm star polymers via copper(I) catalyzed azide-alkyne cycloaddition.
The unique structural and chemical properties of the C60 fullerene and its various derivatives have led to such structures receiving intense attention in the fields of chemistry and materials science.1 In the field of polymer chemistry, various approaches have been utilized for the generation of C60-polymer hybrid materials with various physical properties and morphologies.2 Star-shaped polymers and block copolymers bearing a C60 core are particularly interesting due to their ability to form well-defined microstructures in the solid state.3 The most frequently used method for the synthesis of such C60-based star polymers is the addition of terminal carbanions of polymers prepared via anionic polymerization directly to C60.2a,3 While such a technique works rather well, the severe limitation of applicable monomers as well as the stringent reaction conditions required limits the variety of materials that may be synthesized. Recent trends in polymer chemistry demonstrate that click chemistry has been an attractive route to prepare well-defined polymer conjugates.4 To date, the copper(I) catalyzed azide-alkyne cycloaddition (CuAAC) has been the main representative reaction of the click chemistry philosophy5 due to its high efficiency, accessibility and orthogonality. Thus far, the CuAAC has been utilized in preparing various C60 hexakis adducts6 and C60-polymer conjugates7 by either equipping the fullerene surface with relatively low molecular weight azide or alkyne functionalities.
To the best of our knowledge, there are no reports on the use of the CuAAC for the synthesis of well-defined star polymers bearing a C60 core. It is hypothesized that the larger core will lead to higher efficiency conjugations resulting from a reduction in steric hindrance in the system.
If our hypothesis is confirmed, it would aid in the future design of core molecules for the convergent synthesis of highly branched polymeric systems. As such, we herein report the first examples of well-defined C60-based star polymers prepared by the CuAAC of alkyne-terminated polymers and the recently reported 6-fold azide functionalized hexakis methanofullerene malonate crown-ether 1 (Scheme 1).8
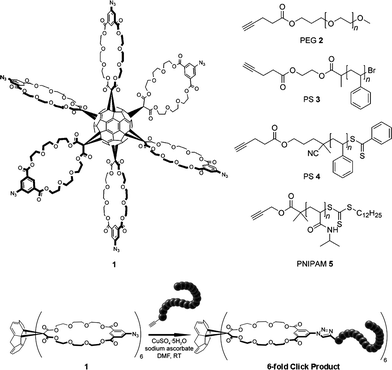 | ||
| Scheme 1 (Top) Structures of the 6-fold azide functionalized hexakis methanofullerene malonate crown-ether 1 and the various alkyne-functionalized polymers synthesized for conjugation, (bottom) a general schematic of the 6-fold polymeric click reaction. | ||
As an initial evaluation of the performance of 1 as a core molecule in the convergent synthesis of star polymers, commercially available poly(ethylene glycol) monomethyl ether (PEG) was equipped with an alkyne moiety via DCC coupling with 4-pentynoic acid (PEG 2 in Scheme 1). The quantitative functionalization of PEG 2 was confirmed by 1H-NMR spectroscopy.
The click reaction between 6 equivalents of PEG 2 with 1 was performed at ambient temperature in DMF solution in the presence of copper(II) sulfate and sodium ascorbate. After stirring for 18 h, the reaction mixture was passed though a short column of neutral alumina to remove the copper catalyst. The solvent was removed by evaporation and the product dried under reduced pressure. The series of molecular weight distributions (MWD) labelled A in Fig. 1 are of the starting materials 1, PEG 2 and the 6-fold click product, denoted here as PEG66. In comparison to the starting materials, the conjugate shows a clear and complete shift to higher molecular weight. The number average molecular weights, Mn, of the respective components are presented in Table 1. It can clearly be observed that the theoretical value of Mn for the six-arm PEG star deviates from the experimentally measured value. This can be explained by the reduced hydrodynamic volume of star-shaped macromolecules in relation to linear analogues of the same molecular weight. However, Radke et al.9 have reported the application of contraction factors that may be used to correct the apparent molecular weight of star polymers (as determined by gel permeation chromatography (GPC)) to a value that reflects the true molecular weight. The contraction factor for a 6-arm star is 1.42, thus multiplying this number with the GPC determined value of Mn approximates, reasonably well, the expected molecular weight of the star. Applying this to the Mn for PEG66 (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 680 g mol−1) gives a corrected value of Mn,corrected = 16590 g mol−1. Upon inspection of the data presented in Table 1, this value is in excellent agreement with the theoretically expected value of Mn,theoretical = 16417 g mol−1. Furthermore, the success of the conjugation was also confirmed by 1H-NMR spectroscopy. These observations, along with the clear disappearance of peaks due to starting materials in the MWD of the 6-fold click product, indicates the efficiency of the CuAAC. Importantly, previous reports on the use of a click approach in the synthesis of star-shaped polymers make use of much smaller and sterically more congested core molecules, bearing the required clickable function.10 It is observed that the efficiency of star formation decreases as the number of branch points on the core increases. Such an observation is primarily due to steric crowding of the core molecule. As more and more arms are added to the core, the accessibility of the remaining reactive sites decreases. In the present case, the core molecule is significantly larger, with the reactive sites for conjugation well separated which results in a reduction of the effect of steric crowding and, thus, the efficiency of star formation is enhanced.
680 g mol−1) gives a corrected value of Mn,corrected = 16590 g mol−1. Upon inspection of the data presented in Table 1, this value is in excellent agreement with the theoretically expected value of Mn,theoretical = 16417 g mol−1. Furthermore, the success of the conjugation was also confirmed by 1H-NMR spectroscopy. These observations, along with the clear disappearance of peaks due to starting materials in the MWD of the 6-fold click product, indicates the efficiency of the CuAAC. Importantly, previous reports on the use of a click approach in the synthesis of star-shaped polymers make use of much smaller and sterically more congested core molecules, bearing the required clickable function.10 It is observed that the efficiency of star formation decreases as the number of branch points on the core increases. Such an observation is primarily due to steric crowding of the core molecule. As more and more arms are added to the core, the accessibility of the remaining reactive sites decreases. In the present case, the core molecule is significantly larger, with the reactive sites for conjugation well separated which results in a reduction of the effect of steric crowding and, thus, the efficiency of star formation is enhanced.
| Compound | M n/g mol−1a | PDI a | M n,corrected/g mol−1b | M n,theo./g mol−1c |
|---|---|---|---|---|
| a Measured via GPC in N,N′-dimethylacetamide (DMAc) (calibrated with PS standards). b Corrected by multiplying GPC Mn values with contraction factor (1.42). c Calculated by the sum of the individual building blocks. d Relative to PS. | ||||
| 1 | 4300 | 1.04 | — | 3943 |
| PEG 2 | 2000 | 1.05 | — | — |
| PEG66 | 11700 |
1.05 | 16590 |
16417 |
| PS 3 | 12200 |
1.19 | — | — |
| PS67 | 55400 |
1.12 | 78670 |
77140 |
| PS 4 | 4200 | 1.06 | — | — |
| PS68 | 21900 |
1.05 | 31100 |
29143 |
| PNIPAM 5 | 10500d |
1.10 | — | — |
| PNIPAM69 | 43100d |
1.08 | 61200 |
66649 |
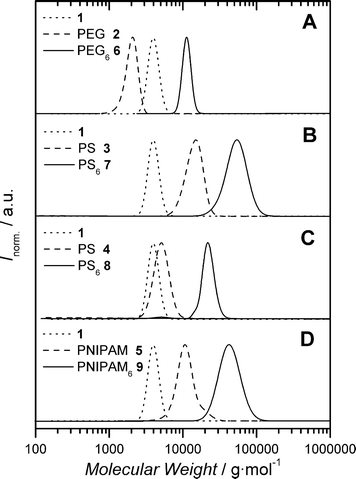 | ||
| Fig. 1 Molecular weight distributions of the various click products in relation to those of the corresponding precursors. | ||
Having established the efficiency of 1 as a core molecule for the convergent synthesis of star-shaped polymers, further alkyne-functionalized polymers were prepared. Both atom transfer radical polymerization (ATRP) and reversible addition fragmentation chain transfer (RAFT) polymerization were used to prepare poly(styrene) (PS 3, 4) with the required functionality (Scheme 1). The presence of the alkyne moiety was verified by 1H-NMR spectroscopy. The click reactions were performed under identical conditions to that of the PEG example, resulting in well-defined star-shaped polymers PS67 and PS68 (Fig. 1, part B and C). The experimental values of Mn presented in Table 1 for both examples are in good agreement with the theoretically expected values.
Recently, Li et al.11 reported the synthesis of C60 coupled to thermoresponsive linear diblock copolymers and homopolymers via the CuAAC. Further examples of a similar nature have also been reported.12 Consequently, we extended our investigation to include the synthesis of star-shaped poly(n-isopropylacrylamide) (PNIPAM). In aqueous media, PNIPAM exhibits a lower critical solution temperature (LCST), implying that a sharp phase transition occurs once a particular temperature is reached. For linear PNIPAM, the LCST is typically 32 °C. Below this temperature, the polymer is water soluble; however upon heating to and above the LCST the chains dehydrate and aggregate which leads to polymer insolubility. This thermoresponsive behaviour of PNIPAM in aqueous media can vary quite substantially with changes in polymer chain end group,13,14 architecture,14,15 concentration16 and molecular weight.17
Alkyne-functionalized PNIPAM 5 was synthesized by RAFT polymerization, the structure of which is presented in Scheme 1. The presence of the alkyne end-group was verified by 1H-NMR spectroscopy. The click reaction of PNIPAM 5 with 1 again proved to proceed very efficiently, as evidenced by the data in Table 1 and the MWD presented in Fig. 1 (part D).
The resulting PNIPAM69 was not at all water soluble at ambient temperature, which can be explained upon inspection of the polymer's structure. PNIPAM69 consists of a highly hydrophobic core with 6 hydrophilic PNIPAM arms radiating outwards. The Z-group of the RAFT agent used in the preparation of PNIPAM 5, the hydrophobic dodecyl chain, is retained in the final PNIPAM69 as the outer chain termini. The presence of hydrophobic end groups in highly branched PNIPAM can lower the LCST such that the polymer is insoluble at ambient temperature,14 thus explaining the present observation.
Interestingly, upon heating a stirred aqueous dispersion of PNIPAM69, the particles were observed to self-flocculate as shown in Fig. 2. It has been reported that while aqueous solutions of PNIPAM turn turbid when heated to above its LCST, it can take as long as 24 h for the chains to fully precipitate.15 It was discovered that by heating the aqueous dispersion of PNIPAM69 more rigorously (>50 °C), the onset of particle flocculation could be induced in the space of minutes. Upon cooling, the particles re-dispersed and the overall flocculation-dispersion process was found to be reversible.
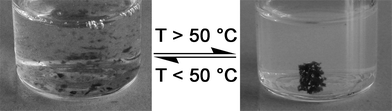 | ||
| Fig. 2 Reversible, thermally induced flocculation of PNIPAM69. | ||
In an attempt to improve the water solubility of PNIPAM69, the hydrophobic dodecyl end-groups were removed by aminolysis (Scheme 2). The success of this transformation was verified by UV/Vis spectroscopy by monitoring the reduction in absorbance at 310 nm (characteristic of the RAFT agent used). A slight decrease in Mn was also observed, which is consistent with the removal of the dodecyl chain termini. Fig. 3 shows the optical transmittance of 3 mg mL −1 aqueous solutions of PNIPAM 5 and the modified PNIPAM610 at various temperatures. As can be clearly observed, the now water soluble PNIPAM610 retains the LCST behaviour of PNIPAM. For a more in depth discussion, the reader is directed to the ESI.†
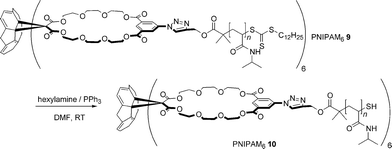 | ||
| Scheme 2 Aminolysis of PNIPAM69. | ||
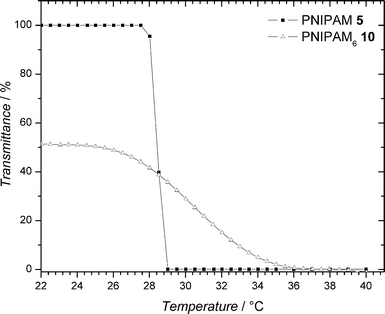 | ||
| Fig. 3 Transmittance vs. temperature for 3 mg mL−1 aqueous solutions of PNIPAM 5 and PNIPAM610 measured at 700 nm. | ||
The present contribution has, for the first time, shown that hexakisazido fullerene derivatives such as 1 may be efficiently used in the synthesis of well-defined, star-shaped polymers via click chemistry. Importantly, we have shown that the use of a large, sterically non-demanding core such as C60 allows for a near flawless star construction even at high molecular weights. In addition, thermoresponsive star-shaped fullerene derivatives have become easily accessible. Further investigations will focus upon the thermoresponsive and aggregation behaviour of such novel materials.
C.B.-K. acknowledges funding from the Karlsruhe Institute of Technology (KIT) within the context of the Excellence Initiative for leading German universities. We gratefully acknowledge the financial support of the Alexander von Humboldt foundation (grant to P.P.) and of the DFG Center for Functional Nanostructures (CFN, Project C5.2).
Notes and references
- M. Prato, J. Mater. Chem., 1997, 7, 1097 RSC.
- (a) F. Giacalone and N. Martin, Chem. Rev., 2006, 106, 5136 CrossRef CAS; (b) H. Wakai, T. Shinno, T. Yamaguchi and N. Tsubokawa, Polymer, 2007, 48, 1972 CrossRef; (c) P. Zhou, G. Q. Chen, H. Hong, F. S. Du, Z. C. Li and F. M. Li, Macromolecules, 2000, 33, 1948 CrossRef CAS.
- (a) B. Schmaltz, M. Brinkmann and C. Mathis, Macromolecules, 2004, 37, 9056 CrossRef CAS; (b) B. Schmaltz, C. Mathis and M. Brinkmann, Polymer, 2009, 50, 966 CrossRef CAS.
- (a) R. A. Evans, Aust. J. Chem., 2007, 60, 384 CrossRef CAS; (b) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952 CrossRef CAS.
- C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987 CrossRef CAS.
- (a) A. Trabolsi, M. Elhabiri, M. Urbani, J. L. D. de la Cruz, F. Ajamaa, N. Solladie, A. M. Albrecht-Gary and J. F. Nierengarten, Chem. Commun., 2005, 5736 RSC; (b) J. Iehl, R. P. De Freitas, B. Delavaux-Nicot and J. F. Nierengarten, Chem. Commun., 2008, 2450 RSC; (c) J. Iehl, R. P. De Freitas and J. F. Nierengarten, Tetrahedron Lett., 2008, 49, 4063 CrossRef CAS.
- W. B. Zhang, Y. Tu, R. Ranjan, R. M. Van Horn, S. Leng, J. Wang, M. J. Polce, C. Wesdemiotis, R. P. Quirk, G. R. Newkome and S. Z. D. Cheng, Macromolecules, 2008, 41, 515 CrossRef CAS.
- P. Pierrat, S. Vanderheiden, T. Muller and S. Bräse, Chem. Commun., 2009, 1748 RSC.
- W. Radke, J. Gerber and G. Wittmann, Polymer, 2003, 44, 519 CrossRef CAS.
- (a) H. F. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 4960 CrossRef CAS; (b) A. J. Inglis, S. Sinnwell, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 4120 CrossRef CAS.
- C. Li, J. Hu, J. Yin and S. Liu, Macromolecules, 2009, 42, 5007 CrossRef CAS.
- (a) A. Tamura, K. Uchida and H. Yajima, Chem. Lett., 2006, 35, 282 CrossRef CAS; (b) G. Zhou, I. I. Harruna, W. L. Zhou, W. K. Aicher and K. E. Geckeler, Chem.–Eur. J., 2007, 13, 569 CrossRef CAS.
- Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275 CrossRef CAS.
- A. P. Vogt and B. Sumerlin, Macromolecules, 2008, 41, 7368 CrossRef CAS.
- R. Plummer, D. J. T. Hill and A. K. Whittaker, Macromolecules, 2006, 39, 8379 CrossRef CAS.
- A. Housni and R. Narrain, Eur. Polym. J., 2007, 43, 4344 CrossRef CAS.
- Y. Xia, X. C. Yin, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2005, 38, 5937 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, NMR, GPC and UV/Vis spectroscopy data and extended discussion section. See DOI: 10.1039/b920806m |
| This journal is © The Royal Society of Chemistry 2010 |