Control of nano/molecular systems by application of macroscopic mechanical stimuli
Katsuhiko
Ariga
*,
Taizo
Mori
and
Jonathan P.
Hill
World Premier International (WPI) Research Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS) and JST, CREST, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: ARIGA.Katsuhiko@nims.go.jp; Fax: +81 29 860 4832; Tel: +81 29 860 4957
First published on 9th August 2010
Abstract
The potential usefulness and importance of nanomaterials are now well recognized. However, currently available synthetic nanomaterials are generally used in their bulk form and control of nanosystems at the nanoscale on demand has not been realized. To solve this problem, the use of macroscopic mechanical stimuli to drive nano/molecular systems is considered to be a key technique. If direct manipulation of nano/molecular systems could be achieved by applying macroscopic mechanical stimuli, we might exert control over nanotechnological systems according to our needs. In this perspective, recent methodologies for controlling nano/molecular systems through application of macroscopic mechanical forces are introduced. Application of mechanical processes is known to affect some molecular association and chemical reactions, causing variation of optical properties, sometimes resulting in self-healing functions or capture and release of molecules under macroscopic mechanical motions. We might be able to realize the great potential of nanoscale and molecular systems by accessing nanoscience and nanotechnology from the macroscopic world.
 Katsuhiko Ariga | Katsuhiko Ariga is principal investigator of World Premier International (WPI) Research Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS). He also works as a director of the Supermolecules group at NIMS and professor at Tokyo University of Science. His major interests are the fabrication of novel nanostructures based on molecular recognition and self-assembly, including Langmuir–Blodgett films, layer-by-layer films, and mesoporous materials. |
 Taizo Mori | Taizo Mori is a postdoctoral researcher in the Supermolecules Group at the National Institute for Materials Science (NIMS). He graduated from the Department of Polymer Chemistry at Kyoto University and obtained his doctorate in 2008. His research interests include supramolecular science, and synthesis of conjugated polymer in liquid crystal and chirality control of molecules. |
 Jonathan P. Hill | Jonathan P. Hill is a senior researcher in the Supermolecules Group at the National Institute for Materials Science (NIMS). Prior to working in NIMS, he occupied postdoctoral positions in Tokyo, University of Karlsruhe (Germany), and Osaka. His research interests include supramolecular science, chemistry of the tetrapyrroles, molecular self-assembly in the bulk state and at interfaces, and the sensing, optical and electronic properties of molecules. |
Accessing nano from macro: learning from Nature
‘Nanoscience’ and ‘nanotechnology’ were formerly technical terms in specialized research areas but they have recently entered everyday use, and the importance of nanomaterials is now well recognized.1–18 However, currently available synthetic nanomaterials are generally used in their bulk form (e.g., as polymer fillers), and control of nanosystems at the nanoscale on demand has not been realized. Thus, nanotechnology might be seen as an immature technology partly because of the lack of means for connecting the nanoscopic world with our activities in the macroscopic world. Integration of the nanoscopic and macroscopic worlds is one of the anticipated breakthroughs for nanotechnology and may bring great benefits to society.Approaches for accessing nano from macro require a means to connect bulk stimuli with molecular level systems. Electronic and photonic stimuli can be respectively regarded as pulses of electrons and photons, which can interact with nano/molecular systems. Similarly, chemical stimuli can be divided at the molecular level into single molecule events. There exist a variety of molecular machines, including motors and tweezers, which have been demonstrated to operate under such stimuli.19–21 In those systems, intramolecular rotations and/or deformations can be stimulated by applying photonic or chemical inputs. In contrast, it is currently challenging to drive nano/molecular systems by applying macroscopic mechanical stimuli since we cannot directly manipulate molecules. However, driving nano/molecular systems by application of macroscopic mechanical stimuli should give rise to many potential methods for connecting macroscopic and nanoscopic systems. In the event of such a connection being made, we might exercise better control over nanoscience and nanotechnology by manipulating nano/molecular systems directly through application of macroscopic mechanical forces.
Examples of how macroscopic forces can be connected to molecular level phenomena are available in biomolecular systems. For instance, humans are constantly exposed to bulk mechanical stresses whose effects must be directly or indirectly converted to molecular events within our bodies. In Nature, evolution has brought about sophisticated mechanisms which bridge the nano and macro length scales. For instance cells can convert a mechanical stimulus into an electrical signal (called mechanotransduction).22 In several mechanosensory systems, a transduction channel can detect deflection of an external structure relative to an internal structure such as deformation of the skin, oscillation of a hair cell bundle, or vibration of a fly's bristle. The probability of a channel opening varies depending on tensional perturbations of the corresponding elements caused by the deflection. Interestingly, such systems have often evolved against some constant background force, i.e., so that mechanotransduction must not be sensitive to a constant external stimuli such as gravity. Adaptation could occur through deformation of the external coupling structure, lengthening of the external or internal anchors, slipping of the internal anchor relative to the cytoskeleton, or changes within the channel.
Similarly, mechanical transduction can be achieved in non-covalent molecular assemblies such as cell membranes.23–25 Increasing tension within a lipid bilayer membrane increases the probability of opening of the membrane channels. A reduction in the occupied area of channels from closed-state to open-state leads to a lower free energy (area–decrease × tension). In one case, a decrease of free energy of ca. 80 pN nm per channel was observed. Channel opening is favoured because of the energetic cost of a hydrophobic mismatch upon thinning of the transmembrane domain during application of lateral tension (see Fig. 1A). Some mechanosensitive channels require asymmetry of the tension gradient across the lipid bilayer for channel opening26,27 so that embedding the channel proteins in symmetric phosphatidylcholine membranes, detergent aggregate, or symmetric lysophosphatidylcholine membrane does not result in channel opening, which can only occur at an asymmetric lysophosphatidylcholine membrane. The resulting open state is highly dynamic, supporting a water-filled pore of at least 2.5 nm. Apart from open/closed control of channels, binding of ligand molecules to receptor proteins can be tuned by application of mechanical stimuli. As shown in Fig. 1B, mechanical forces trigger exposure of protein binding sites through changes in protein conformation (case a) or folding (case b).28,29
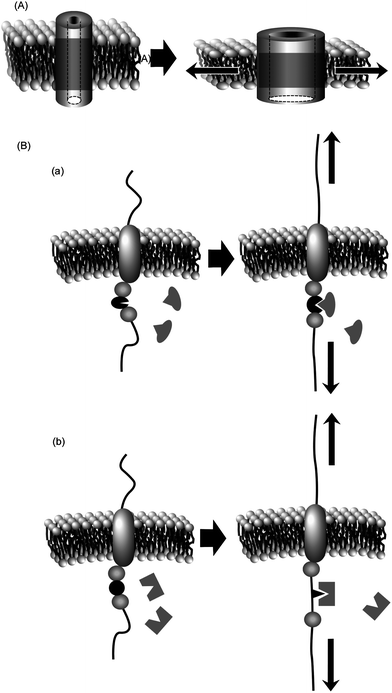 | ||
| Fig. 1 Mechanical control of protein functions within membrane media: (A) control of channel opening; (B) control of receptor activity. | ||
From the available biochemical systems that connect nano- and macroscale effects, it can be appreciated that great complexity of design is not necessarily a prerequisite in the structures of analogous synthetic systems. Control of nanosystems by application of macroscopic mechanical stimuli should become possible if molecules and their assemblies are appropriately designed. In this article, we summarize recent advances in mechanical control of nano and molecular phenomena as a perspective of one of the future directions of nanoscience and nanotechnology. We illustrate how nanoscience and nanotechnology can be accessed physically from the macroscopic world according to three categories, physico-chemical properties, chemical events, and bio-related functions. Also included are some approaches in which macroscale phenomena are stimulated through molecular action at the nanoscale.
Control of physico-chemical properties
Structural features of materials can be modified by mechanical stresses, perhaps leading to alternation between two physico-chemical states. In particular, control of photonic function has been investigated since the optical properties of materials can be strongly dependent on arrangement, orientation, and state of aggregation of chromophores. These factors can also be influenced through mechanical stretching or deformation of the materials.Yamazaki and coworkers investigated intramolecular excitation energy transfer within bichromophoric compounds containing a naphthalene donor and anthracene acceptor linked through a methylene spacer.30 These compounds were then embedded in stretched and unstretched polymer matrices. When incorporated in the stretched polymer (poly(vinyl alcohol), PVA), the bichromophoric compounds were forced to take a conformation elongated in the stretching direction. Analyses of picosecond time-resolved fluorescence spectra and fluorescence decay kinetics revealed a systematic variation depending on the length of the interchromophore spacer. In contrast, the energy transfer rates in non-stretched polymer (poly(methyl methacrylate), PMMA) films were faster than those in stretched PVA films and systematic changes depending on spacer length could not be detected. In the latter matrix, the molecules probably take up a folded conformation. Dynamic control of optical properties of dye molecules through stretching of matrix polymers has also been reported. Marawske et al. investigated correlations between the impact of an external mechanical force on a molecular framework of fluorophores and the resultant variation in their fluorescence properties using an oligo(paraphenylene vinylene) derivative with a twisted molecular backbone embedded in a thin matrix of poly(vinyl chloride) (PVC).31 Stretching the matrix by applying uniaxial force simultaneously caused three major optical effects due to changes in chromophore geometry, i.e., an increase in fluorescence anisotropy, a decrease in the fluorescence lifetime, and an increase in the emission energy. Calculations revealed that a mechanical stress of 6.9 nN leads to a 0.2 ns shortening of fluorescence lifetime. Application of 0.2 − 0.55 nN per molecule resulted in a 35 ps shortening of the lifetime. This system is expected to provide optical sensors that can monitor local mechanical stress in transparent samples down to the single-molecule level.
Liquid crystalline materials are also sensitive to mechanical stimuli. Sagara and Kato reported a shear-induced liquid-crystalline phase transition of a pyrene derivative, which in turn induces a change in photoluminescence wavelength (Fig. 2).32 Variation in the assembled structures of luminescent groups at the phase transition causes changes in the photoluminescence properties of the materials. The subject materials exhibit yellow photoluminescence in the cubic phase, which becomes blue-green following a phase transition. This change can also be triggered by mechanical shearing of the cubic liquid crystalline phase. Segmented columnar structures in the cubic phase are formed by hydrogen bonding between amide groups of adjacent molecules resulting in formation of π-stacked structures of the emissive cores, which exhibit yellow excimer fluorescence emission. Application of shear-force induces the phase transition with formation of non-segmented columnar structures. The emissive cores are then not overlapped in this structure preventing excimer formation of the pyrene moieties. The blue-green photoluminescence of the shear-induced columnar phase was thus observed because of changes in the self-assembly structure.
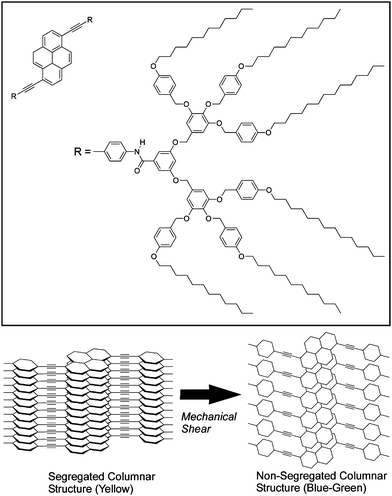 | ||
| Fig. 2 Control of stacking of liquid crystalline molecules through mechanical shearing. | ||
Ito, Sawamura, and coworkers demonstrated mechanochromic luminescence behaviour of [(C6F5Au)2(μ-1,4-diisocyanobenzene)].33 Photoluminescence in solid state samples of this compound undergoes a drastic variation upon grinding and exposure of the solid to solvents or solvent vapour. The mechanical action of grinding transforms the blue luminescent microcrystalline powder into a metastable amorphous phase, where aurophilic interactions are responsible for the lower energy emission. Exposure of the ground sample to a solvent induces rearrangements of the amorphous phase into the more stable crystalline phase through a partial dissolution and recrystallization process. Using this phenomenon, they demonstrated writing of yellow figures on a blue background by macroscopic scratching. Araki and coworkers reported luminescence changes of a solid amide-substituted tetraphenylpyrene derivative based on a pressure-dependency of molecular packing.34 Quadruple amide hydrogen bonds result in formation of a well defined columnar assembly in the solid state of the blue-emitting B-form. Application of pressure to the B-form caused disruption of the columnar structure yielding the G-form with a poorly ordered molecular packing. The G-form possessed a different luminophore arrangement and consequently showed greenish luminescence. This system is rewritable, i.e., writing by pressing or grinding to the green-emitting state and erasing by heating to the blue-emitting state, and is easy to reproduce. These materials might find a wide variety of applications such as in optical recording media or in strain- or pressure-sensing systems.
Control of nanostructure by using mechanical force has also been reported. Xu and coworkers reported conversion of mechanical stimuli into photonic output in praseodymium-doped ceramics. In that case, red-light emission was produced upon compression of the ceramic materials.35 These materials may find application in multifunctional sensors or devices. Fudouzi and Sawada demonstrated that a silicone rubber sheet could exhibit tunable and reversible structural color due to mechanical strain.36 The sheets were made from a thin layer of cubic close-packed (ccp) colloidal particles embedded in poly(dimethylsiloxane) (PDMS) elastomer. The sheet-stretching induced a decrease of the lattice distance of ccp (111) planes, which was manifested as a blue shift of absorption wavelength. In this system, a 20% extension of the sheet induced a 30 nm shift in the wavelength of the absorption maximum. The Bragg's diffraction peak of the photonic rubber sheet could be tuned linearly as a function of mechanical strain upon elastic deformation. Accordingly, the color of the PDMS sheet varied from red to green. The material investigated is expected to find uses in applications such as optical sensing of strain or colour decoration by strain.
Control of chemical events including self-healing and molecular recognition
Deformation of materials could induce local changes such as chemical reaction or molecular/coordination complexation. This feature would lead to mechanical control of phenomena including self-healing and on-demand molecular recognition.Moore and coworkers demonstrated force-induced covalent-bond activation and materials' color changes (Fig. 3)37,38 in mechanophore-linked elastomeric and glassy polymers. Spiropyran units underwent a reversible ring-opening reaction under tensile stress, realizing locally visualization of the mechanochemical reaction. Mechanoresponsive polymeric materials were synthesized by linking spiropyran moieties into the chains of bulk polymers or by using spiropyran as crosslinkers. Applying mechanical forces reversibly transformed the closed, colorless spiropyran form to a highly colored, planar merocyanine structure through rupture of the spiro group carbon-oxygen (C–O) bond. The bond cleavage was induced by a force of 2 nN per molecule, which is somewhat lower than values for C–C bond cleavage at 2.3 − 13.4 nN. The spiropyran mechanophore functions as a molecular force sensor, providing visible detection and mapping of mechanical stresses within bulk polymeric materials. Although the mechanism of chemical bond cleavage and formation are not clear, this system might serve as a new mechanophore building unit, possibly imparting polymeric materials with desirable functionalities ranging from damage sensing to fully regenerative self-healing.
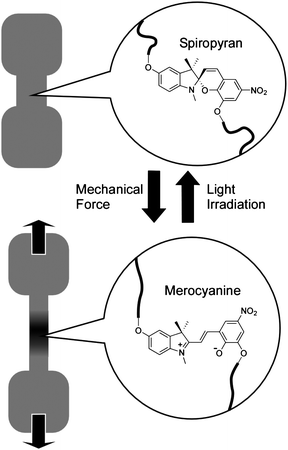 | ||
| Fig. 3 Control by mechanical stretching of isomerization between spiropyran and merocyanine attached to polymer chains. | ||
In other work, Leibler and coworkers proposed a non-covalent design for the preparation of mechanically self-healing polymers.39 They synthesized materials composed of molecules that associate through hydrogen bonds to form both chains and cross-links. The fatty diacids used in their design as hydrogen-bond-forming components were derived from natural renewable resources (vegetable oils). They are liquid at room temperature and do not crystallize, forming glasses instead. The materials can contain variable amounts of triacids. A mixture of fatty diacid and triacid was condensed first with diethylene triamine and then reacted with urea giving a mixture of oligomers equipped with complementary hydrogen bonding groups: amidoethyl imidazolidone, di(amidoethyl) urea and diamido tetraethyl triurea. The materials showed recoverable extensibility up to several hundred percent and little creep under load. When water rather than dodecane was used as plasticizer, the glass transition was lowered and an extension of 500% could be completely reversed within a few seconds of release of the applied strain, much as in conventional rubbers, and stretched samples could be maintained under stress for about ten hours without appreciable creep. More surprisingly, even when broken or cut, the materials could be repaired simply by bringing together fractured surfaces, which self-heal at room temperature.
As an example of control of molecular complexation, rapid and selective assembly of an acceptor–donor–acceptor (ADA) complex using electron rich crown ether and electron deficient naphthalene diimide derivatives through mechanical grinding of a binary mixture was demonstrated by Liu and coworkers.40 Charge transfer complexes containing columnar alternating ADA stacks could be readily tuned to modulate electronic, optical, and ferroelectric properties. These complexes were usually obtained in solution through solvophobic or ion-binding interactions. However, formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complex through a solid–to–solid mechanical grinding process was also demonstrated. This is an interesting example of control of supramolecular complex formation by application of an external macroscopic mechanical process.
:2 complex through a solid–to–solid mechanical grinding process was also demonstrated. This is an interesting example of control of supramolecular complex formation by application of an external macroscopic mechanical process.
At interfacial media, supramolecular phenomena often exhibit features quite different from those in the bulk phase. For example, molecular recognition efficiencies are known to be greatly enhanced at the air–water interface relative to those in bulk water.41–47 Therefore, use of interfaces for mechanical control of nano/molecular phenomena is also a significant subject. In addition, dynamic interfaces should be considered as a medium for more sophisticated mechanical control of nano/molecular phenomena. For instance, a Langmuir monolayer spread on a liquid surface at the air–water interface can be visibly compressed and expanded in the interfacial plane. Nanoscopic and/or molecular-level changes perpendicular to the film can occur stimulated by macroscopic deformation of the film. Use of a dynamic interface might be one of the best approaches for controlling nano/molecular systems through macroscopic mechanical motions.48
As shown in Fig. 4A, a steroid cyclophane molecule with a cyclic core consisting of a 1,6,20,25-tetraaza[6.1.6.1] paracyclophane connected to four steroid moieties (cholic acid) through a flexible L-lysine spacer was used as a monolayer component.49–53 This operates as a molecular machine in the binding of a guest molecule through variation between planar and cavity-forming conformations of the cyclophane. Binding of guest molecules dissolved in the water subphase was investigated using the fluorescent dye, 6-(p-toluidino)naphthalene-2-sulfonate (TNS). Because fluorescence of the TNS molecule is largely quenched in a highly polar aqueous medium but exhibits strong emission when trapped in a hydrophobic cavity, detection of fluorescence at the air–water interface enabled detection of guest binding by the steroid cyclophane. The observed fluorescence changes upon binding of the guest to the steroid cyclophane were synchronous with the bulk monolayer motions, where compression of the monolayer to surface pressures in the range 25 to 50 mN m−1 induces efficient guest binding. Compression and expansion of the monolayer resulted in respective increase or decrease in fluorescence intensity of TNS. Therefore, binding and release of the guest molecule could be repeated by compression and expansion of the monolayer. This is a clear demonstration of guest binding and release by application of bulk mechanical forces.
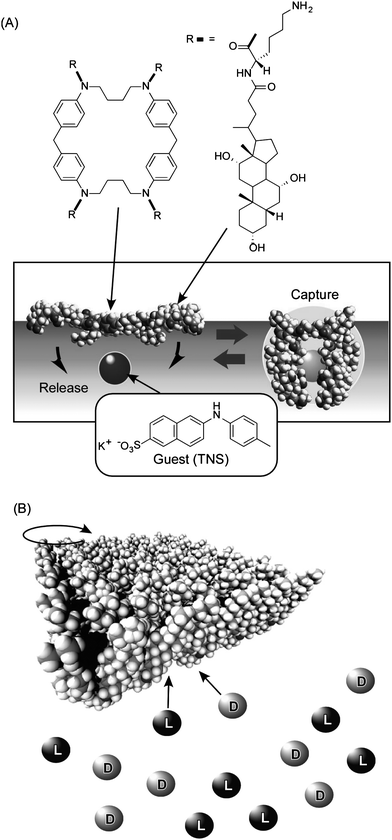 | ||
| Fig. 4 (A) Molecular capture and release by steroid cyclophane molecules in Langmuir monolayer by mechanical compression and expansion of the monolayer. (B) Chiral discrimination of guests by molecular twisting of cholesteryl-substituted cyclen depending on surface pressure. | ||
In another example, we employed a cholesteryl-substituted cyclen complex host molecule, and demonstrated inversion of enantioselectivity in molecular recognition controlled by macroscopic lateral pressure applied to the monolayer at the air–water interface (Fig. 4B).54,55 The octacoordinate sodium complex of cholesteryl-substituted cyclen has two possible quadruple helicate structures. Helicity is influenced by the chirality of the side arms especially when ordered or aggregated at the supramolecular level. Accommodation of chiral guest molecules within the hydrophobic cavities also affects the helicity. Binding of amino acids in aqueous subphase to the monolayer was examined. The binding values of D-leucine are always greater than those of L-leucine at all surface pressures investigated, indicating that the monolayer of cholesteryl-substituted cyclen has a stronger interaction with D-leucine. In sharp contrast, the values of L-valine are smaller than those of D-valine at low surface pressure but exceed them at 22–23 mN m−1. Therefore, chiral recognition in the monolayers of cholesteryl-substituted cyclen with valine changes from the D- to L-form upon compression. This is a clear example of tuning of chiral discrimination by bulk mechanical force. Remarkably, a small difference in the amino acid structure resulted in differing selectivities, a feature comparable to the delicate functions of enzymes and receptors in living organisms.
Control of bio-related functions
Further challenges exist in mechanical control of more complicated nano- and micro-systems. In particular, control of biological systems by applying artificial macroscopic forces is an attractive research target.Sheetz and coworkers demonstrated that mechanical stretching of single cytoplasmic proteins can activate binding of other molecules using magnetic tweezers in combination with total internal reflection fluorescence spectroscopy and atomic force microscopy.56 Application of physiologically relevant forces caused stretching of single talin rods that exposed cryptic binding sites for vinculin. Thus, in the talin–vinculin system, molecular mechanotransduction can occur by protein binding after exposure of the buried binding sites. Vogel and coworkers similarly investigated structures and functions of fibronectin under application of mechanical forces.57,58
Mechanical operation of biological systems has also been investigated by combining biomaterials and nanostructures. Yoshida and coworkers demonstrated direct visualisation of the rotary motion of a single molecule of F1-ATPase.59 They attached a fluorescent actin filament to the gamma-subunit of F1-ATPase as a marker in order to visualize its rotatory motion directly. Addition of ATP induced the filament rotation (more than 100 revolutions) in a counterclockwise direction from the membrane side with concurrent conversion of ATP to ADP. Control of this molecular motion was also realised by application of external macroscopic motions through immobilization of magnetic nanoparticles on the F1-ATPase (Fig. 5).60 In this molecular motor, the reverse rotation driven by a proton gradient is known to be responsible for ATP synthesis in biological systems. It was also demonstrated that reverse rotation of the ATPase upon external stimuli resulted in ATP production, i.e., chemical synthesis using mechanical energy. A magnetic bead was attached to the γ-subunit of isolated F1 on a glass surface and was rotated using an electromagnet. Rotation in the appropriate direction resulted in the appearance of ATP in the medium, which was detected by using the luciferase–luciferin reaction. This work is a fine demonstration of control of chemical synthesis by an external vectorial force.
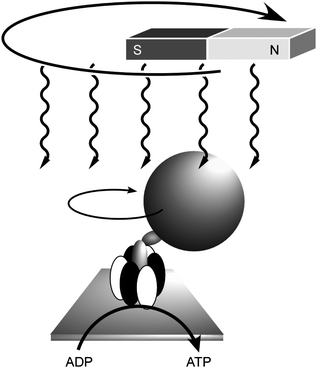 | ||
| Fig. 5 Control of rotation of ATPase by the macroscopic motion of magnetic fields with concurrent ATP synthesis. | ||
Manipulation of large biomolecules using small sharp tips has also been occasionally successfully applied. For example, Morita and Shingyogi investigated protein sliding in microtubules through mechanical bending of flagella attached to a small glass tip.61 Arakawa, Ikai and coworkers investigated mechanical properties of an individual protein molecule through direct force measurements by pulling the protein using the tip of an atomic force microscope (AFM).62 In other work, Shigekawa et al. successfully shifted a molecular unit of a polyrotaxane structure.63 They prepared polyrotaxanes containing several cyclodextrin molecules on a poly(ethyleneglycol) thread. Observation of the polyrotaxane by scanning tunneling microscope (STM) revealed sliding of individual cyclodextrin molecules on the polymer thread. It is believed that the mechanical action of the STM tip induced sliding of the cyclodextrin unit.
Mechanically-controlled switching of enzymatic reactions can be realized by combining naturally occurring enzymes with supramolecular thin films. Of the known thin film media, layer-by-layer (LbL) assemblies64–72 are known to ‘softly’ accommodate enzyme molecules while maintaining their function by permitting the requisite freedom of motion,73–81 in contrast to other condensed films such as Langmuir–Blodgett (LB) films.82–88 Voegel and coworkers reported mechanical control of enzymes (Alkaline phosphatase, ALP) embedded in an LbL assembly (Fig. 6).89,90 The assembled film consisted of a primary polyelectrolyte multilayer stratum loaded with ALP which was capped with a second polyelectrolyte layer acting as a mechanically sensitive nanobarrier. ALP enzymes in the capped unstretched film did not perform catalytic conversion of a fluorescein diphosphate substrate. Under stretching, enzymes became accessible to the substrates.and at a critical stretching degree of 70%, ALP enzymes were exposed so that biocatalysis was permitted and hydrolysis of fluorescein diphosphate molecules to fluorescein and phosphate ions commenced. An immediate strong enhancement of green fluorescence was observed in the very top part of the LbL film. A high production rate of fluorescein molecules and phosphate ions led to their release in solution and also to a large local increase of their concentration in or close to the barrier inducing feedback inhibition of the enzymes. The concentration of phosphate ions produced during hydrolysis should also locally increase within the barrier, resulting in feedback inhibition of the enzymes and explains why rinsing of the LbL film with buffer reactivates enzymes for further catalysis. Unstretching the LbL films forces remasking of the ALP enzymes by the polyelectrolytes, i.e. catalysis was switched off. This cycle makes a good model for control of exposure of active sites as has been observed in biological mechanotransduction systems.
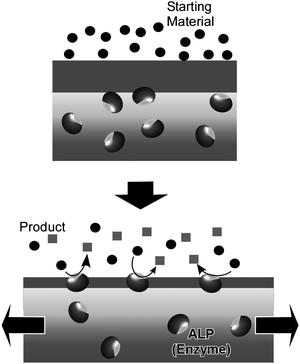 | ||
| Fig. 6 Control by mechanical stretching of activity of enzymes embedded in an LbL assembly. | ||
Contrasting challenges: controlling macroscale phenomena using nanoscopic forces
For connecting nanoscopic and macroscopic systems in mechanical function, an approach involving control of macroscopic mechanical phenomena using nano/molecular events is another important challenge. This approach relies on forcing macroscopic phenomena by amplification and/or accumulation of nanoscopic and molecular events. Since this approach might provide insight into molecular design for connecting nano/molecular systems and the macroscopic world, we will briefly introduce some examples.Ichimura and coworkers demonstrated manipulation of orientation and motion of a thick fluid layer through control of orientation of a molecular monolayer. Known as a command surface, alignment of a layer of a liquid crystal can be triggered by photo-isomerization of surface-bound molecules (Fig. 7A).91 It should be emphasized that the alignment of a thick liquid crystal layer (a macroscopic object) is determined only by the photochromic monolayer (nanoscopic structures). They also reported control of the lateral motion of a liquid droplet on a self-assembled monolayer (SAM) bearing a photoresponsive unit (Fig. 7B).92 Photoirradiation of the azobenzene-functionalized monolayer with UV light (365 nm) resulted in the formation of ca. 90% cis isomer, which has a larger dipole moment than the trans isomer. Photoirradiation of the cis-rich surface with blue light (365 nm) in turn transformed the cis isomer into the trans isomer, and thus, the surface energy was returned to its lower level. Irradiation of the SAM surface with UV light or blue light respectively resulted in enhanced or reduced surface wetting by a droplet of olive oil. The gradient of the surface energy between the advancing and receding edges of the droplet could be constantly maintained by moving the light beam. Motion of the droplet could be continued upon irradiation of blue light with graded intensity. This demonstrates amplification of nanoscopic/molecular effects in order that they be observed at the macroscale.
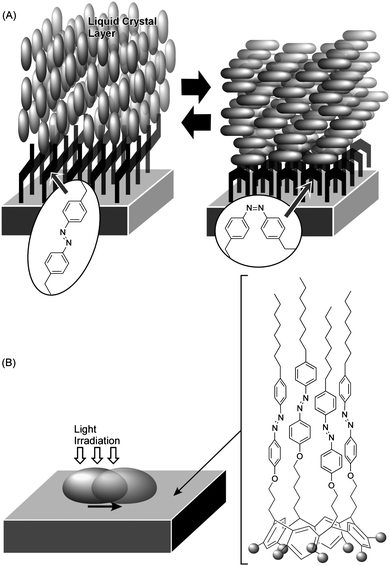 | ||
| Fig. 7 Control of motion of large fluidic objects by photo-isomerization of surface monolayer: (A) control of liquid crystal orientation at a command surface; (B) control of lateral movement of a liquid droplet. | ||
The importance of the accumulation of nanoscopic actions to the connection between nanoscopic and macroscopic phenomena can also be recognized in the following two examples. Ikeda and co-workers showed that a single film of a liquid crystal network containing an azobenzene chromophore can be repeatedly and precisely bent in a selected direction by using linearly polarized light.93 Exposure of the film to 366 nm polarized light induced film bending in the direction of light polarization. In turn, irradiation of the film with visible light of a wavelength longer than 540 nm returned the film to its initial flat state. Thus, accumulation of molecular level activity was used to induce deformation of objects that are visible to the naked eye.
Mechanical molecular machines including molecular rotors and molecular shuttles have also been investigated.94 In these systems, molecular motions can be controlled and mechanical motions at the molecular level can also be converted to macroscopic mechanical events. For example, Stoddart and coworkers demonstrated that accumulation of motion of molecular machines can actually lead to deformation of macroscopic objects. In their approach, numerous molecular shuttle units were immobilized on the surface of an AFM cantilever (Fig. 8).95 Shifting the position of the shuttle unit along the molecular axis resulted in molecular-level torsions, which accumulated tension at the AFM cantilever surface causing bending of the much larger cantilever. A mean molecular force of 14 to 21 pN caused cantilever deformation of 35 to 50 nm. Similarly, motion of molecular shuttles can be converted into liquid motion (wettability) as demonstrated by Zhang and coworkers.96 Modulation of surface pressure was realized by motions of molecular shuttles, which were vertically aligned at the air–water interface, as reported by Stoddart, Ho and coworkers.97
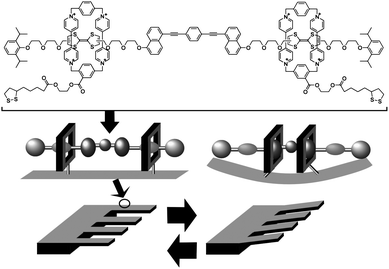 | ||
| Fig. 8 Conversion of molecular shuttle motion into deformation of a macroscopic object. | ||
Future challenges
In this short perspective, we have described recent research aimed at controlling nanoscopic and molecular systems by using macroscopic mechanical stimuli. The examples presented illustrate how access to and control of molecules and nanostructures can be exercised through macroscopic manipulation. These approaches can be categorised as a new family of nanotechnological devices that can be mechanically operated from the macroscale. Mechanical stimuli are general and ubiquitous in macroscopic machines but are not well understood or exploited in devices at the nanoscopic and molecular scales. However, application of appropriately designed molecules or molecular assemblies or use of dynamic interfaces such as Langmuir monolayers enables us to easily compress, expand, and deform the nano/molecular systems. Also, combining an appropriately deformable material such as a gel could also lead to development of unique stimuli-responsive systems. However, ambiguous dispersion of functional molecular units at interfaces and within the materials results in uncertainty of stimuli–response relationships between external mechanical forces and molecular motion. Therefore, the development of strategies for the precise control of molecular arrangement, including intermolecular orientation and packing, will be a critical issue in further progress on control of nano/molecular systems by macroscopic mechanical forces.Since humans exist and operate ostensibly at the macroscopic scale, advanced nanosystems and molecular mechanisms seem to be disconnected from our daily activities. Ultrasmall objects might not appear to be of direct use in our daily activities so that connection between our macroscale activities and nano/molecular systems for nanotechnological purposes requires well considered design concepts. As described in this perspective, chemical sciences including molecular design and synthesis, materials chemistry, and colloid/interface chemistry play crucial roles in realizing approaches to nano/molecular systems from the macroscopic world.
Acknowledgements
This work was partly supported by World Premier International Research Center Initiative (WPI Initiative), MEXT, Japan and Core Research for Evolutional Science and Technology (CREST) program of Japan Science and Technology Agency (JST), Japan.Notes and references
- C. R. Martin, Chem. Mater., 1996, 8, 1739 CrossRef CAS.
- S. Förster and M. Antonietti, Adv. Mater., 1998, 10, 195 CrossRef.
- A. C. Grimsdale and K. Müllen, Angew. Chem., Int. Ed., 2005, 44, 5592 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS.
- K. Ariga, J. P. Hill, M. V. Lee, A. Vinu, R. Charvet and S. Acharya, Sci. Technol. Adv. Mater., 2008, 9, 014109 CrossRef.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664 RSC.
- S. Acharya, U. K. Gautam, T. Sasaki, Y. Bando, Y. Golan and K. Ariga, J. Am. Chem. Soc., 2008, 130, 4594 CrossRef CAS.
- N. Tanaka, Sci. Technol. Adv. Mater., 2008, 9, 014111 CrossRef.
- M. Sathish, K. Miyazawa, J. P. Hill and K. Ariga, J. Am. Chem. Soc., 2009, 131, 6372 CrossRef CAS.
- N. A. Kotov, J. O. Winter, I. P. Clements, E. Jan, B. P. Timko, S. Campidelli, S. Pathak, A. Mazzatenta, C. M. Lieber, M. Prato, R. V. Bellamkonda, G. A. Silva, N. W. S. Kam, F. Patolsky and L. Ballerini, Adv. Mater., 2009, 21, 3970 CrossRef CAS.
- S. Acharya, D. D. Sarma, Y. Golan, S. Sengupta and K. Ariga, J. Am. Chem. Soc., 2009, 131, 11282 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752 CrossRef CAS.
- N. Pradhan, S. Acharya, K. Ariga, N. S. Karan, D. D. Sarma, Y. Wada, S. Efrima and Y. Golan, J. Am. Chem. Soc., 2010, 132, 1212 CrossRef CAS.
- E. Ruiz-Hitzky, M. Darder, P. Aranda and K. Ariga, Adv. Mater., 2010, 22, 323 CrossRef CAS.
- K. Ariga, X. Hu, S. Mandal and J. P. Hill, Nanoscale, 2010, 2, 198 RSC.
- B. Baroli, J. Pharm. Sci., 2010, 99, 21 CrossRef CAS.
- K. Ariga, Q. Ji, J. P. Hill and A. Vinu, J. Inorg. Organomet. Polym. Mater., 2010, 20, 1 CrossRef CAS.
- S. Mandal, M. V. Lee, J. P. Hill, A. Vinu and K. Ariga, J. Nanosci. Nanotechnol., 2010, 10, 21 CrossRef CAS.
- V. Balzani, M. Gomez-Lopez and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405 CrossRef CAS.
- A. Harada, Acc. Chem. Res., 2001, 34, 456 CrossRef CAS.
- K. Kinbara and T. Aida, Chem. Rev., 2005, 105, 1377 CrossRef CAS.
- P. G. Gillespie and R. G. Walker, Nature, 2001, 413, 194 CrossRef CAS.
- B. Martinac and O. P. Hamill, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4308 CrossRef CAS.
- P. A. Janmey and D. A. Weitz, Trends Biochem. Sci., 2004, 29, 364 CrossRef CAS.
- A. W. Orr, B. P. Helmke, B. R. Blackman and M. A. Schwartz, Dev. Cell, 2006, 10, 11 CrossRef CAS.
- E. Perozo, D. M. Cortes, P. Sompornpisut, A. Kloda and B. Martinac, Nature, 2002, 418, 942 CrossRef CAS.
- B. Martinac, J. Cell Sci., 2004, 117, 2449 CrossRef CAS.
- A. F. Oberhauser, P. E. Marszalek, H. P. Erickson and J. M. Fernandez, Nature, 1998, 393, 181 CrossRef CAS.
- Y. Sawada and M. P. Sheetz, J. Cell Biol., 2002, 156, 609 CrossRef CAS.
- M. Hasegawa, S. Enomoto, T. Hoshi, K. Igarashi, T. Yamazaki, Y. Nishimura, S. Speiser and I. Yamazaki, J. Phys. Chem. B, 2002, 106, 4925 CrossRef CAS.
- S. Marawske, D. Dörr, D. Schmitz, A. Koslowski, Y. Lu, H. Ritter, W. Thiel, C. A. M. Seidel and R. Kühnemuth, ChemPhysChem, 2009, 10, 2041 CrossRef CAS.
- Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175 CrossRef CAS.
- H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044 CrossRef CAS.
- Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520 CrossRef CAS.
- X. Wang, C.-N. Xu, H. Yamada, K. Nishikubo and X.-G. Zheng, Adv. Mater., 2005, 17, 1254 CrossRef CAS.
- H. Fudouzi and T. Sawada, Langmuir, 2006, 22, 1365 CrossRef CAS.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68 CrossRef CAS.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977 CrossRef CAS.
- G. Koshkakaryan, L. M. Klivansky, D. Cao, M. Snauko, S. J. Teat, J. O. Struppe and Y. Liu, J. Am. Chem. Soc., 2009, 131, 2078 CrossRef CAS.
- X. Cha, K. Ariga, M. Onda and T. Kunitake, J. Am. Chem. Soc., 1995, 117, 11833 CrossRef CAS.
- M. Onda, K. Yoshihara, H. Koyano, K. Ariga and T. Kunitake, J. Am. Chem. Soc., 1996, 118, 8524 CrossRef CAS.
- X. Cha, K. Ariga and T. Kunitake, J. Am. Chem. Soc., 1996, 118, 9545 CrossRef CAS.
- M. Sakurai, H. Tamagawa, Y. Inoue, K. Ariga and T. Kunitake, J. Phys. Chem. B, 1997, 101, 4810 CrossRef CAS.
- H. Tamagawa, M. Sakurai, Y. Inoue, K. Ariga and T. Kunitake, J. Phys. Chem. B, 1997, 101, 4817 CrossRef CAS.
- K. Ariga and T. Kunitake, Acc. Chem. Res., 1998, 31, 371 CrossRef CAS.
- K. Ariga, A. Kamino, X. Cha and T. Kunitake, Langmuir, 1999, 15, 3875 CrossRef CAS.
- K. Ariga, M. V. Lee, T. Mori, X.-Y. Yu and J. P. Hill, Adv. Colloid Interface Sci., 2010, 154, 20 CrossRef CAS.
- K. Ariga, Y. Terasaka, D. Sakai, H. Tsuji and J. Kikuchi, J. Am. Chem. Soc., 2000, 122, 7835 CrossRef CAS.
- K. Ariga, T. Nakanishi, Y. Terasaka, H. Tsuji, D. Sakai and J. Kikuchi, Langmuir, 2005, 21, 976 CrossRef CAS.
- K. Ariga, T. Nakanishi, J. P. Hill, Y. Terasaka, D. Sakai and J. Kikuchi, Soft Matter, 2005, 1, 132 RSC.
- K. Ariga, T. Nakanishi and J. P. Hill, Soft Matter, 2006, 2, 465 RSC.
- K. Ariga, T. Nakanishi, Y. Terasaka and J. Kikuchi, J. Porous Mater., 2006, 13, 427 CrossRef CAS.
- T. Michinobu, S. Shinoda, T. Nakanishi, J. P. Hill, K. Fujii, T. N. Player, H. Tsukube and K. Ariga, J. Am. Chem. Soc., 2006, 128, 14478 CrossRef CAS.
- K. Ariga, T. Michinobu, T. Nakanishi and J. P. Hill, Curr. Opin. Colloid Interface Sci., 2008, 13, 23 CrossRef CAS.
- A. del Rio, R. Perez-Jimenez, R. Liu, P. Roca-Cusachs, J. M. Fernandez and M. P. Sheetz, Science, 2009, 323, 638 CrossRef CAS.
- M. Gao, D. Craig, O. Lequin, I. D. Campbell, V. Vogel and K. Schulten, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14784 CrossRef CAS.
- M. L. Smith, D. Gourdon, W. C. Little, K. E. Kubow, R. A. Eguiluz, S. Luna-Morris and V. Vogel, PLos Biol., 2007, 5, 2243 CAS.
- H. Noji, R. Yasuda, M. Yoshida and K. Kinosita Jr., Nature, 1997, 386, 299 CrossRef CAS.
- H. Itoh, A. Takahashi, K. Adachi, H. Noji, R. Yasuda, M. Yoshida and K. Kinosita Jr., Nature, 2004, 427, 465 CrossRef CAS.
- Y. Morita and C. Shingyoji, Curr. Biol., 2004, 14, 2113 CrossRef CAS.
- K. Mitsui, K. Nakajima, H. Arakawa, M. Hara and A. Ikai, Biochem. Biophys. Res. Commun., 2000, 272, 55 CrossRef CAS.
- H. Shigekawa, K. Miyake, J. Sumaoka, A. Harada and M. Komiyama, J. Am. Chem. Soc., 2000, 122, 5411 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232 CrossRef CAS.
- P. T. Hammond, Adv. Mater., 2004, 16, 1271 CrossRef CAS.
- K. Ariga, J. P. Hill and Q. Ji, Phys. Chem. Chem. Phys., 2007, 9, 2319 RSC.
- Q. Ji, M. Miyahara, J. P. Hill, S. Acharya, A. Vinu, S. B. Yoon, J.-S. Yu, K. Sakamoto and K. Ariga, J. Am. Chem. Soc., 2008, 130, 2376 CrossRef CAS.
- K. Ariga, A. Vinu, Q. Ji, O. Ohmori, J. P. Hill, S. Acharya, J. Koike and S. Shiratori, Angew. Chem., Int. Ed., 2008, 47, 7254 CrossRef CAS.
- S. Srivastava and N. A. Kotov, Acc. Chem. Res., 2008, 41, 1831 CrossRef CAS.
- Q. Ji, S. B. Yoon, J. P. Hill, A. Vinu, J.-S. Yu and K. Ariga, J. Am. Chem. Soc., 2009, 131, 4220 CrossRef CAS.
- J. A. Lichter, K. J. Van Vliet and M. F. Rubner, Macromolecules, 2009, 42, 8573 CrossRef CAS.
- Q. Ji, S. Acharya, J. P. Hill, A. Vinu, S. B. Yoon, J.-S. Yu, K. Sakamoto and K. Ariga, Adv. Funct. Mater., 2009, 19, 1792 CrossRef CAS.
- Y. Lvov, K. Ariga, I. Ichinose and T. Kunitake, J. Am. Chem. Soc., 1995, 117, 6117 CrossRef CAS.
- M. Onda, Y. Lvov, K. Ariga and T. Kunitake, J. Ferment. Bioeng., 1996, 82, 502 CrossRef CAS.
- M. Onda, Y. Lvov, K. Ariga and T. Kunitake, Biotechnol. Bioeng., 1996, 51, 163 CrossRef.
- M. Onda, K. Ariga and T. Kunitake, J. Biosci. Bioeng., 1999, 87, 69 CrossRef CAS.
- H. Ai, S. A. Jones and Y. M. Lvov, Cell Biochem. Biophys., 2003, 39, 23 CrossRef CAS.
- Y. Wang, A. S. Angelatos and F. Caruso, Chem. Mater., 2008, 20, 848 CrossRef CAS.
- K. Ariga, J. P. Hill and Q. Ji, Macromol. Biosci., 2008, 8, 981 CrossRef CAS.
- Q. He, Y. Cui and J. Li, Chem. Soc. Rev., 2009, 38, 2292 RSC.
- K. Ariga, Q. Ji and J. P. Hill, Adv. Polym. Sci., 2010, 229, 51.
- Y. Okahata, T. Tsuruta, K. Ijiro and K. Ariga, Langmuir, 1988, 4, 1373 CrossRef CAS.
- Y. Okahata, T. Tsuruta, K. Ijiro and K. Ariga, Thin Solid Films, 1989, 180, 65 CrossRef CAS.
- K. Ariga, T. Nakanishi and T. Michinobu, J. Nanosci. Nanotechnol., 2006, 6, 2278 CrossRef CAS.
- X. Chen, S. Lenhert, M. Hirtz, N. Lu, H. Fuchs and L. Chi, Acc. Chem. Res., 2007, 40, 393 CrossRef CAS.
- D. R. Talham, T. Yamamoto and M. W. Meisel, J. Phys.: Condens. Matter, 2008, 20, 184006 CrossRef.
- S. Acharya, A. Shundo, J. P. Hill and K. Ariga, J. Nanosci. Nanotechnol., 2009, 9, 3 CrossRef.
- S. Acharya, J. P. Hill and K. Ariga, Adv. Mater., 2009, 21, 2959 CrossRef CAS.
- D. Mertz, J. Hemmerlé, J. Mutterer, S. Ollivier, J.-C. Voegel, P. Schaaf and P. Lavalle, Nano Lett., 2007, 7, 657 CrossRef CAS.
- D. Mertz, C. Vogt, J. Hemmerlé, J. Mutterer, V. Ball, J.-C. Voegel, P. Schaaf and P. Lavalle, Nat. Mater., 2009, 8, 731 CrossRef CAS.
- K. Ichimura, Chem. Rev., 2000, 100, 1847 CrossRef CAS.
- K. Ichimura, S.-K. Oh and M. Nakagawa, Science, 2000, 288, 1624 CrossRef CAS.
- Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72 CrossRef CAS.
- Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, B. H. Northrop, H.-R. Tseng, J. O. Jeppesen, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. D. Solares, W. A. Goddard, C.-M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745 CrossRef CAS.
- P. Wan, Y. Jiang, Y. Wang, Z. Wang and X. Zhang, Chem. Commun., 2008, 5710 RSC.
- T. J. Huang, H.-R. Tseng, L. Sha, W. Lu, B. Brough, A. H. Flood, B.-D. Yu, P. C. Celestre, J. P. Chang, J. F. Stoddart and C.-M. Ho, Nano Lett., 2004, 4, 2065 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |